Article Text

Abstract
Thiopurines are often the mainstay of treatment for many patients with inflammatory bowel disease. As such, a general understanding of the evidence behind their use and of their metabolism is extremely useful in clinical practice. This review gives a practical overview of thiopurine metabolism, the importance of thiopurine S-methyltransferase testing prior to the start of therapy and the monitoring of thioguanine nucleotide levels while on treatment, guiding a personalised approach to optimising thiopurine therapy.
- AZATHIOPRINE
- 6-MERCAPTOPURINE
- INFLAMMATORY BOWEL DISEASE
- THIOPURINE METHYLTRANSFERASE
- ADVERSE DRUG REACTIONS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- AZATHIOPRINE
- 6-MERCAPTOPURINE
- INFLAMMATORY BOWEL DISEASE
- THIOPURINE METHYLTRANSFERASE
- ADVERSE DRUG REACTIONS
Introduction
Inflammatory bowel disease (IBD) is a chronic inflammatory disorder of the gut, divided into two main diseases, ulcerative colitis (UC) and Crohn's disease (CD).
Over the last decade there has been a shift in paradigm of the treatment of IBD with a ‘top down’ or ‘rapid step-up’ approach aimed at altering the natural history early in the course of the disease. Newer drugs, especially biologics, are being used for the treatment of IBD, and existing drugs such as thiopurines are being used more effectively by guiding individual dosing according to pharmacogenetic data and monitoring of drug metabolite levels.
Sixty per cent of patients with IBD receive thiopurines (azathioprine (AZA), mercaptopurine (MP) and to a lesser extent tioguanine) with proven efficacy in maintaining steroid-free remission.1 Although the evidence for the use of thiopurines in UC is not as strong as in patients with CD, it has been demonstrated that in patients with steroid-dependent UC, 53% achieved steroid-free remission on AZA compared with 21% on aminosalicylates.2 Therefore, thiopurines are recommended once a patient has required two courses of steroids within a year.3 Thiopurines are commenced earlier in CD, where aminosalicylates play little or no role, and have been demonstrated to reduce the need for surgery by 40%.4 The SONIC (Study of Biologic and Immunomodulator Naive Patients in CD) trial study showed the benefit of thiopurines in addition to anti-tumour necrosis factor (TNF) agents in the treatment of CD, most likely through reduced formation of antidrug antibodies against anti-TNF agents.5
Up to a third of patients have to stop thiopurines due to side effects, the main concerns being leucopenia (1.3–12.6%), hepatotoxicity (4%), pancreatitis (3%) and gastric intolerance (1.3–6%).6 ,7 Thiopurine S-methyltransferase (TPMT) activity influences the incidence of adverse effects, particularly bone marrow toxicity, however, there is no association with hepatotoxicity or pancreatitis.8
Thiopurine metabolism
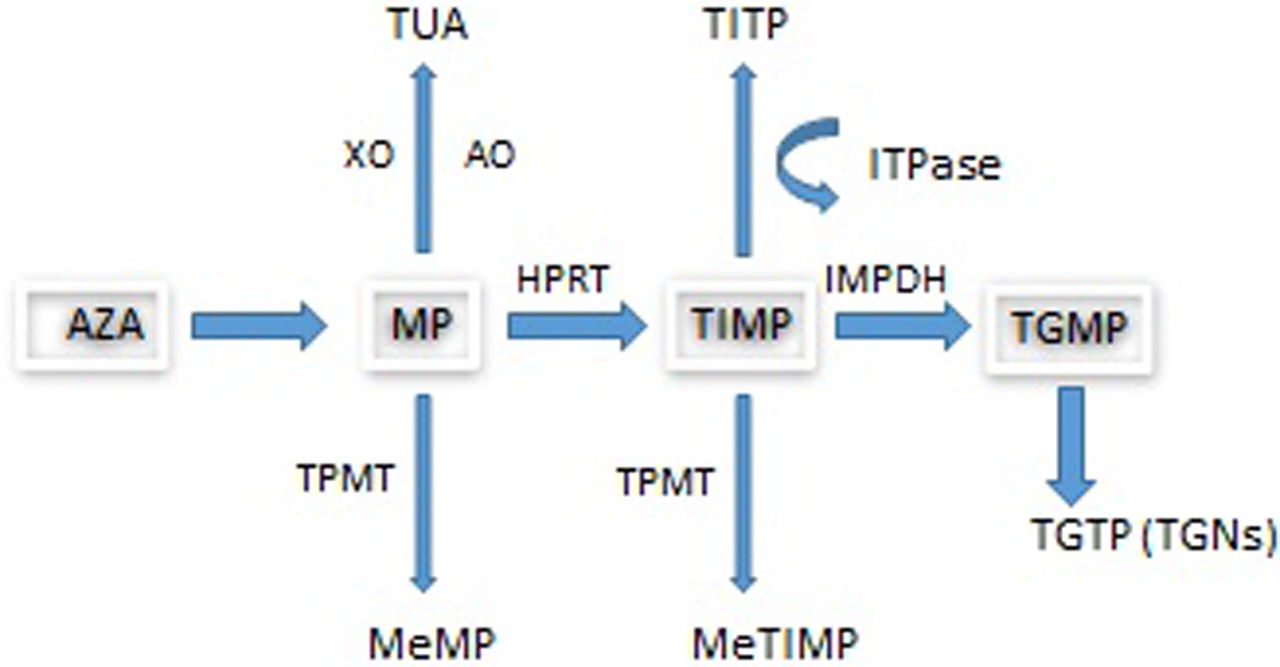
The prodrug AZA is converted non-enzymatically by biogenic thiols, including glutathione, to MP with the release of methyl-4-nitro-5-imidazole. Following uptake by transporters, MP undergoes metabolism by three competing pathways (see figure 1) to form the active metabolite, thioguanine nucleotides (TGNs) which function as rogue nucleic acids, disrupting the DNA replication of the most rapidly dividing cells such as activated T cell lymphocytes where TGN concentrations have been found to be higher and where genes involved in T cell immunity have been shown to be downregulated by AZA.9
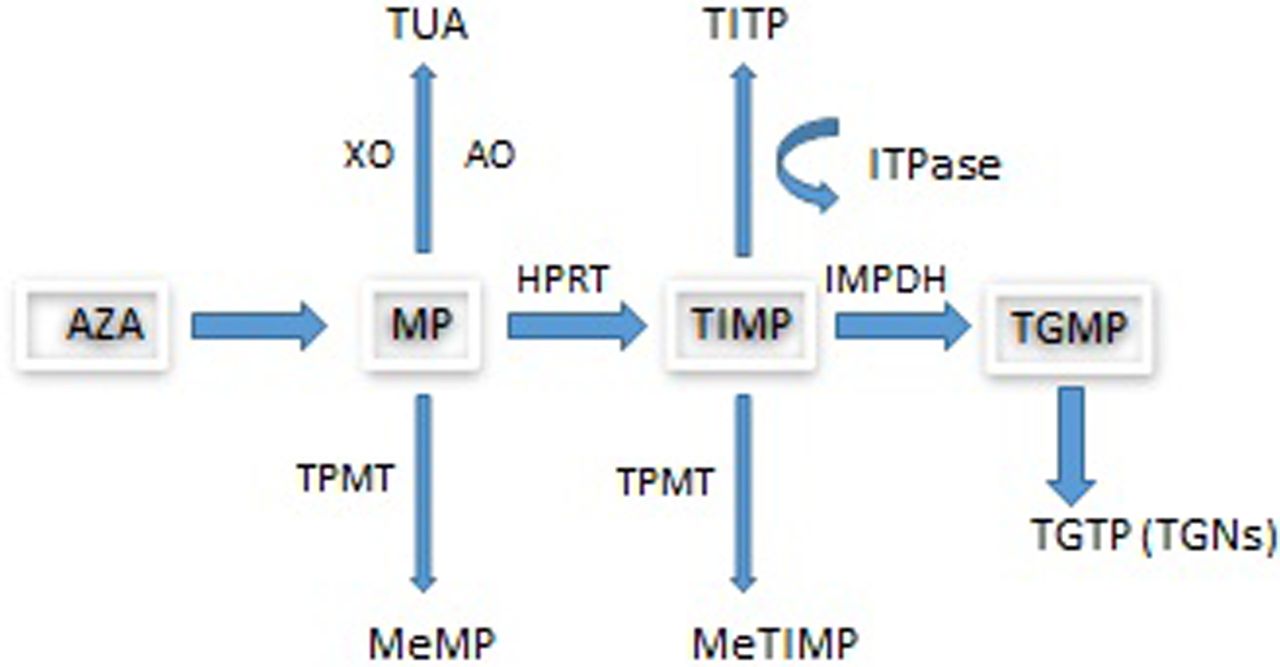
Azathioprine metabolism. AO, aldehyde oxidase; AZA, azathioprine; HPRT, hypoxanthine phosphoribosyltransferase; IBD, inflammatory bowel disease; IMPDH, inosine monophosphate dehydrogenase; ITPase, inosine triphosphatase; MeMP, methylmercaptopurine; MeTIMP, methylthioinosine monophosphate; MP, mercaptopurine; TGMP, thioguanine monophosphate; TGN, thioguanine nucleotide; TGTP, thioguanine triphosphate; TIMP, thioinosine monophosphate; TITP, thioinosine triphosphate; TPMT, thiopurine S-methyltransferase; TUA, thiouric acid; XO, xanthine oxidase. The metabolism of MP involves three competing pathways: the first being degradation to TUA which is then excreted, the second is through methylation by TPMT into MeMP, and the third is the breakdown of MP into TIMP catalysed by HPRT. TIMP is then further metabolised via IMPDH into TGMP. Kinases convert this into the TGNs. Approximately 15–20% of patients with IBD demonstrate hypermethylation when treated with thiopurines. This means that during thiopurine metabolism, methylated thiopurine metabolites are preferentially produced instead of TGNs.
Thiopurine S-methyltransferase
Methylation of MP by TPMT is a critical step in thiopurine metabolism. It was first noted in the 1980s that differences in TPMT activity help account for the variability in tolerance to thiopurines. Rare and common genetic polymorphisms influence enzyme function resulting in a trimodal population distribution of activity. In Caucasians, complete TPMT deficiency occurs in 1 in 300 individuals. Carriers of a deficiency-associated allele (heterozygotes) have around 50% enzyme activity and occur at an approximate frequency of 1 in 10 of the population.10 The majority of cases of TPMT deficiency (∼95%) are associated with three alleles, TPMT*2, TPMT*3A and TPMT*3C.11 The frequency of these variants depends on ethnicity with TPMT*3A being more common in Caucasian populations.12
Individuals with complete TPMT deficiency who receive standard doses of AZA or MP are highly likely to develop severe and potentially fatal myelosuppression. There is a small case series of patients with IBD treated with around 10% of standard doses, all of whom tolerated this well.13 Heterozygotes are also at risk of toxicity at standard doses but this is prevented by initiation at 50% of the conventional dose. Hence, recommended practice is to check TPMT levels prior to starting therapy and adjusting the dose according to TPMT status.
Ultrahigh TPMT (>40 pmol/mg Hb/h) is associated with a skewed drug metabolism in a selection of patients where MP is preferentially metabolised to methylmercaptopurine (MeMP) resulting in lower TGNs which in turn are associated with a poorer clinical response and side effects. This preferential metabolism to MeMP or ‘shunting’ is known as hypermethylation.14
TPMT phenotyping by enzyme assay is generally preferred to genotyping, as the assay will detect all completely deficient patients, irrespective of whether the patient has a rare or common variant TPMT genotype. Other factors influencing TPMT activity, for example red cell age, also result in discordance between genotype and phenotype, particularly in the high carrier range.15
Thioguanine nucleotides
Although important, TPMT polymorphisms account for only 10% of overall thiopurine toxicity.16 Indeed, 50–75% of all patients developing leucopenia have a normal TPMT and only 3% of hypermethylators have ultrahigh TPMT.17 Measuring TGNs provides a summary of epigenetic and genetic factors influencing thiopurine metabolism and, together with measurement of MeMP, offers a means for therapeutic drug monitoring.
Measurement of TGNs and MeMP in red blood cells (RBCs) has been shown to be clinically useful after steady state is reached at 4–6 weeks.18 Meta-analyses suggest that a therapeutic range of TGN between 235 pmol/8×108 and 450 pmol/8×108 RBCs correlates best with a good clinical response.19 ,20 In addition, there is evidence that TGN monitoring in IBD is associated with improved outcomes with a negative correlation between RBC TGNs and disease activity.21 A treatment strategy using TGNs to determine optimal dosing resulted in improved outcomes in 90% of patients compared with 33% in those not guided by TGNs.18
Hypermethylation
Approximately 15–20% of patients with IBD demonstrate hypermethylation when treated with thiopurines.16 The usual definition of hypermethylation is a ratio of MeMP to TGN of >11. In this setting, subtherapeutic TGNs risk a poor response to therapy. Moreover, MeMP >5700 pmol/8×108 RBCs results in a higher risk of hepatotoxicity.20
Allopurinol is a xanthine oxidase inhibitor that prevents the breakdown of thiopurines into thiouric acid (TUA). It was designed to increase the bioavailability of MP by preventing degradation to TUA. The combination of low dose thiopurine and 100 mg of allopurinol (LDTA) corrects hypermethylation in patients who have failed therapy due to hepatotoxicity or who have had a poor response to treatment in association with subtherapeutic TGNs.14 ,22 When using the combination, the administered dose of AZA or MP is reduced to 25–50% of the standard monotherapy dose.23
Allopurinol is a safe drug with the main side effects including rashes and gastrointestinal (GI) symptoms but it can rarely (1:100 000) cause life-threatening toxic epidermal necrolysis.24
A practical guide to thiopurine treatment
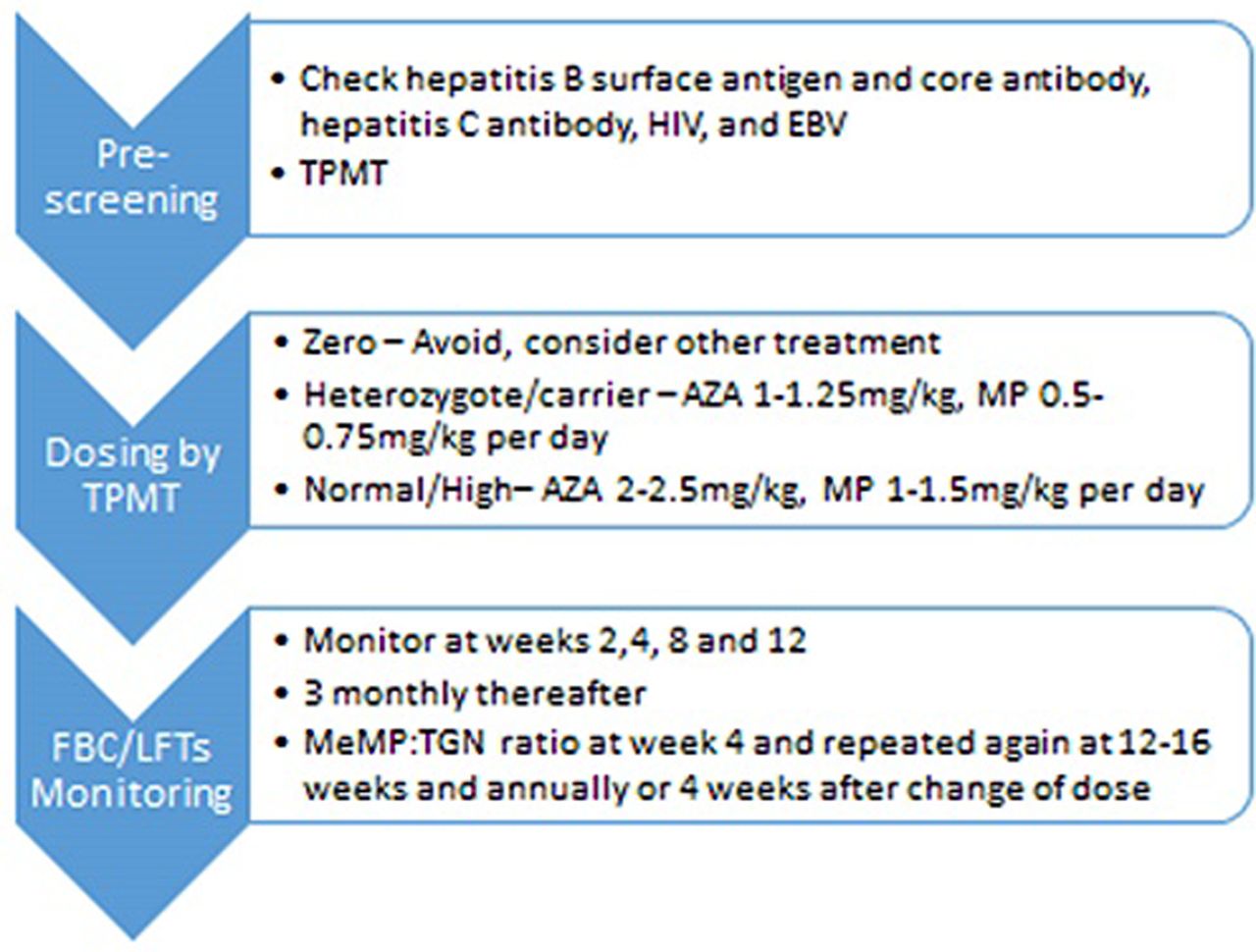
Prior to commencing thiopurines, patients should be screened to help prevent opportunistic infections as per European guidelines (see table 1). In addition, the pneumococcal vaccination is given before starting treatment and the influenza vaccine annually.25 Patients are advised that while on treatment they should not have ‘live’ vaccines. The more common ‘live’ vaccines include yellow fever; measles, mumps and rubella; and BCG (tuberculosis vaccine). The IBD passport is a good source of information for both patients and prescribers on foreign travel with IBD.26
Screening prior to thiopurine treatment25
Epstein-Barr virus (EBV) seronegativity in patients commencing thiopurines has long been a concern and it was identified in 2002 that treatment in this setting was associated with a small increased risk of EBV-positive lymphomas.27 More recently in the CESAME observational cohort, thiopurine treatment had an HR of 5.28 for developing lymphoproliferative disorders with EBV infection being a further risk factor.28 The risk remains small and as yet, avoidance of these drugs in seronegative individuals is not advised. European guidelines suggests considering anti-TNF monotherapy as opposed to combination therapy with thiopurines.25
Optimising clinical response
Measuring TGNs has been proven to improve clinical outcomes.18 We check TGNs in all patients who have symptoms or active disease. We also check TGNs 4 weeks after starting thiopurines or following a change in the dose (see figure 2). The onset of response from thiopurines is around 16 weeks so if being prescribed as monotherapy, bridging with corticosteroids or an alternative therapy should be considered.29

The pathway for commencement of thiopurines and monitoring. AZA, azathioprine; EBV, Epstein-Barr virus; MeMP, methylmercaptopurine; MP, mercaptopurine; FBC, full blood count; LFTs, liver function tests; TGN, thioguanine nucleotide; TPMT, thiopurine S-methyl transferase.
Where TGNs are subtherapeutic, increasing the dose by between 25 mg and 50 mg may be enough for TGNs to reach the therapeutic range. If patients are hypermethylating, and TGNs subtherapeutic, we recommend either switching to LDTA or dose splitting. The rationale behind dose splitting is that the reduced dose of thiopurine is below the optimum substrate affinity for TPMT. In a review of 20 patients with baseline MeMP levels greater than 7000 pmol/8×108 RBCs, dose splitting resulted in a significant decrease in MeMP levels without reducing TGNs.30
Managing side effects
There are a number of different strategies employed when addressing adverse effects experienced on thiopurines which are discussed below (see figure 3).
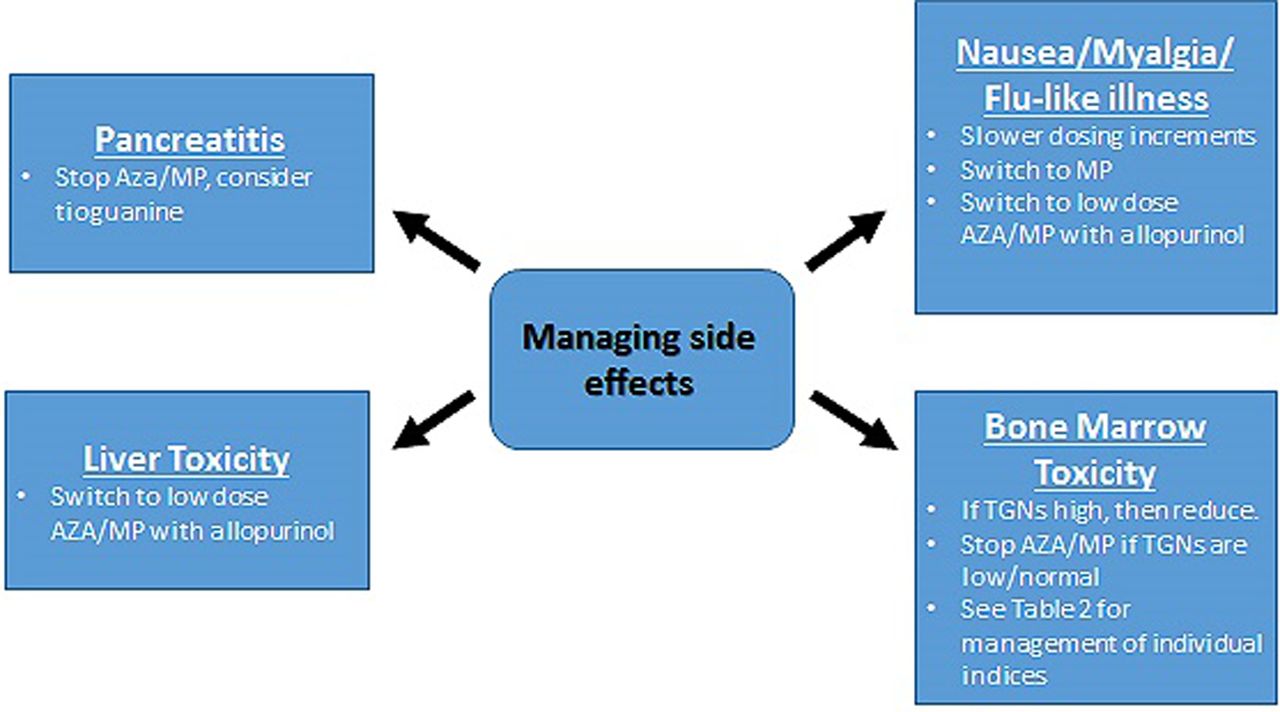
Management of adverse side effects in the context of MeMP:TGN monitoring. AZA, azathioprine; MeMP, methylmercaptopurine; MP, mercaptopurine; TGN, thioguanine nucleotides.
For side effects other than pancreatitis, switching to LDTA achieves clinical remission in 78% of patients and has been found to reduce levels of MeMP while increasing TGNs to within therapeutic range.24 The lowest available doses of AZA and MP come in a liquid form at 10 mg/mL and 20 mg/mL, respectively, both of which are off-licence. For logistical reasons we advise patients who need to take the lowest dose to take 1.5 mL/1 mL of AZA alternate days or halve a 25 mg tablet of MP (12.5 mg).
Gastrointestinal side effects
Nausea is common with thiopurines. Changing the timing of the dose from morning to later in the day or at night can sometimes alleviate this. Switching to MP is well recognised to circumvent some cases of AZA induced nausea.31 A further option is to increase the dose of thiopurine gradually over 2–4 weeks rather than start at the maximum dose. Finally, switching to LDTA has been proven to successfully circumvent nausea in over a half of patients.24
Bone marrow toxicity
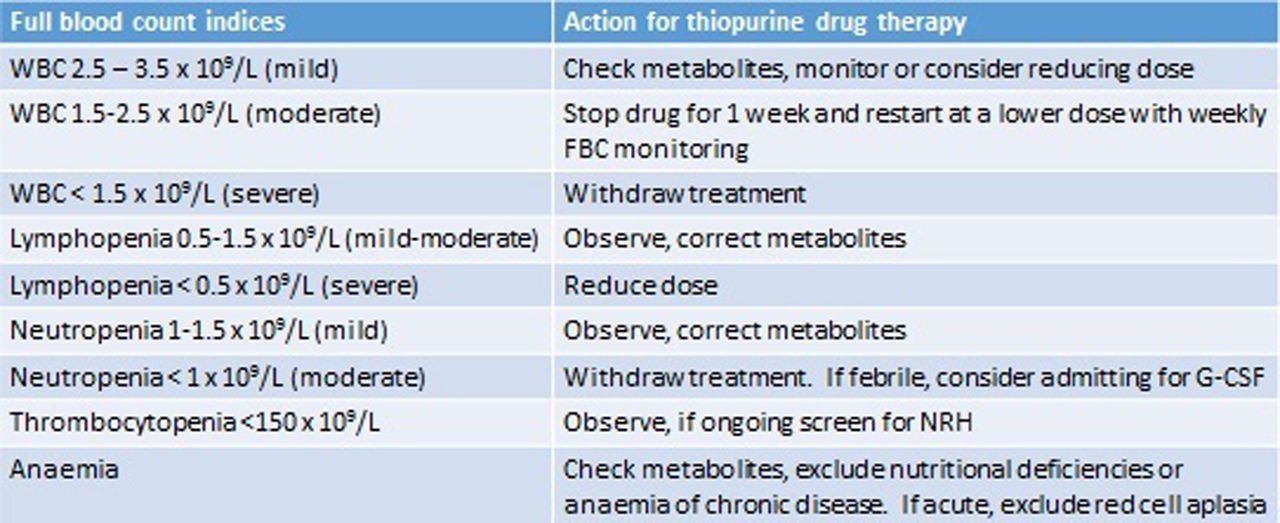
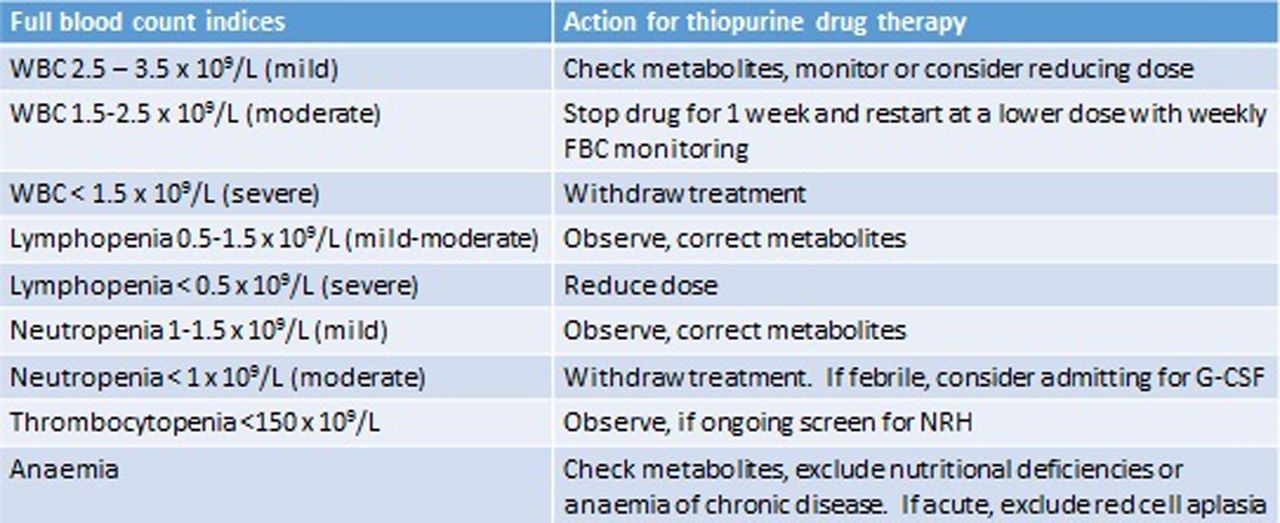
Myelosuppression has been associated with low TPMT activity and with MeMP >11 450 pmol/8×108 RBCs.32 In this scenario, we adopt the same approach to that of hypermethylation and hepatotoxicity and switch to LDTA. Where leucopenia occurs in the absence of hypermethylation, our practice is to alter dosing according to relevant blood count parameters (see figure 4). In the case of lymphopenia, adjustment is debateable, as studies suggest patients with lymphopenia are not at increased risk of infections.33
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The management of deranged full blood count (FBC) indices in patients on thiopurines. G-CSF, granulocyte colony stimulating factor; NRH, nodular regenerative hyperplasia; WBC, white blood cell.
Hepatotoxicity
Four per cent of patients experience thiopurine related hepatotoxicity. If this occurs the drug should be stopped until the liver function tests have normalised followed by commencing LDTA. MeMP levels >5700 pmol/8×108 RBCs have been associated with a threefold increase in the risk of hepatotoxicity.20 However, a proportion of patients develop hepatotoxicity in the absence of hypermethylation. Interestingly, in these patients, hepatotoxicity is also successfully circumvented in most cases by switching to LDTA.
Pancreatitis
In the 3% of patients who develop pancreatitis, thiopurines are immediately stopped. Reports of successful rechallenge or switch to MP have been published, but, in general, the risk of repeat pancreatitis is high and we do not therefore recommend rechallenge with AZA or MP in this setting. Tioguanine, however, at doses of 20–40 mg, is an alternative to AZA and MP, which successfully avoids pancreatitis and is associated with reported clinical remission rates as high as 79% at 12 months.34
Other considerations
The use of thiopurines as concomitant therapy alongside biologics has been shown to reduce antidrug antibody formation.4 The therapeutic range for this is not known. Recent studies have suggested that aiming for lower TGNs in this setting is equally efficacious.35–37
There are good safety data on the use of thiopurines in pregnancy and around the time of conception for women and men and their use is supported by European guidelines.38 ,39 There are however no safety data on the use of allopurinol in pregnancy. We advise the continued use of thiopurines throughout pregnancy after careful discussion with the patient. We avoid starting allopurinol during pregnancy and would advise other treatments in situations where allopurinol would normally be used.
Most centres consider withdrawal after 5 years of treatment if the patient is in clinical remission. Treatment for over 4 years has been shown to increase the risk of non-melanoma skin cancer and lymphoma particularly in those patients over the age of 50 years.40 There is also an association between the long-term use of thiopurines, especially tioguanine, and nodular regenerative hyperplasia. Where thiopurines are withdrawn, 1 year clinical relapse rates are 23% in CD and 12% in UC.41
Conclusion
Thiopurines continue to be the mainstay of therapy for CD and moderate to severe UC. Thiopurine metabolite monitoring in addition to TPMT measurement successfully guides optimised therapy, reducing treatment failure from adverse effects or lack of clinical efficacy and is a classic example of the benefits of personalised medicine. These benefits are clearly evident in the treatment of IBD but should also be relevant across many other disciplines in which thiopurines are used as standard therapy.
References
Footnotes
Contributors Both BW and EJ wrote the manuscript with both MA-H and AM reviewing the biochemistry sections. PI and JS reviewed and supervised the clinical sections.
Competing interests None declared.
Provenance and peer review Not commissioned; internally peer reviewed.