Article Text

Abstract
Hereditary bowel tumours are usually part of a distinct syndrome which require management of both intestinal and extra-intestinal disease. Polyposis syndromes include: Familial adenomatous polyposis, MUTYH-associated polyposis, Serrated polyposis syndrome, Peutz-Jeghers syndrome, Juvenile polyposis syndrome and PTEN-hamartomatous syndromes. Of all colorectal cancers (CRC), 5%–10% will be due to an underlying hereditary CRC syndrome. Diagnosis and management of polyposis syndromes is constantly evolving as new scientific and technological advancements are made with respect to identifying causative genes and increased sophistication of endoscopic therapy to treat polyps. This, in addition to data yielded from meticulous record-keeping by polyposis registries has helped to guide management in what are otherwise relatively rare conditions. These data help guide clinical management of patients and their ‘at-risk’ relatives. Diagnosis is both genetic where possible but clinical recognition is key in the absence of an identifiable causative gene. Furthermore, some syndromes can overlap which can additionally complicate diagnosis. The principle goals of polyposis management are first to manage and treat the presenting patient and then to identify ‘at-risk’ patients, through screening and predictive genetic testing, endoscopic surveillance to allow therapy and guide surgical prophylaxis. Due to the complexity of diagnosis and management, patients and their families should be referred to a genetics centre or a polyposis registry where dedicated management can take place.
- familial adenomatous polyposis
- polyposis
Statistics from Altmetric.com
Summary box
What is already known about this subject?
Hereditary bowel cancer syndromes are responsible for approximately 5% of bowel cancers.
Polyposis syndromes are rare hereditary multisystem disorders which require life-long specialist surveillance.
Operative planning and surveillance management of adenomatous polyposis syndromes relies on specialist knowledge of phenotypic presentations and germline mutations.
What are the new findings?
Increasing occurrence of gastric adenoma in Familial adenomatous polyposis (FAP).
The occurrence of metachronous cancers in serrated polyposis is less than previously thought.
The Juvenile polyposis syndrome -hereditary haemorrhagic telangiectasia overlap in patients with SMAD4 mutations requires specialised cardiac and vascular assessment.
Preimplantation genetic diagnosis (PGD) of embryos can be offered to all patients for family planning where the genetic mutation is known (except for serrated polyposis).
How might it impact clinical practice in the foreseeable future?
PGD offers the option of having a child without inheriting the disease; this will reduce theincidence of polyposis syndromes.
Gastric surveillance in patients with FAP will become routine but further management guidelines are required.
For serrated polyposis, segmental resections when required may be sufficient in the future (pending further published data).
Introduction
Hereditary syndromes are responsible for approximately 5% to 10% of all colorectal cancers (CRC). In the UK, a diagnosis or suspicion of a hereditary syndrome should trigger a referral to a regional clinical genetics centre.
The management of the polyposis patient requires a multidisciplinary team approach delivered by specialised centres such as a dedicated polyposis registry, and will necessitate provision of clinical care to other members of the family by virtue of the inherited nature of the disease. In the UK, there is only one dedicated polyposis registry (St Mark’s Hospital, London). A detailed family history is required to identify at-risk relatives. In turn, these individuals require genetic counselling. The field has evolved significantly since the early 1990s largely due to advancements in genetic technologies; identification of mutations has significantly impacted the management of polyposis patients and families.
Knowledge of the germline mutation may predict and guide management in some conditions. When a germline mutation is not detected in a family, then endoscopic surveillance may be required over time to clinically determine whether the patient develops the polyposis syndrome.
Chemoprevention to delay or prevent surgery in the gastrointestinal (GI) tract is an attractive proposition but unfortunately there are no data that support its routine use in these conditions. Thus, the cornerstone of management is endoscopic surveillance and polypectomy and where appropriate, prophylactic surgery.
This review will describe the practical management of the following polyposis syndromes.
Adenomatous polyposis syndromes
Familial adenomatous polyposis (FAP)
MUTYH-associated adenomatous polyposis (MAP)
Serrated polyposis syndrome (SAP)
Hamartomatous polyposis
Peutz-Jeghers syndrome (PJS)
Juvenile polyposis syndrome (JPS)
PTEN Hamartoma tumour syndromes
Familial adenomatous polyposis
FAP is caused by a mutation in the APC gene located on chromosome 5q211 and has an incidence of 1: 10 000. It has an autosomal dominant pattern of inheritance; although 20% of mutations arise de novo. Clinically it manifests with up to hundreds or thousands of adenomas in the colorectum. CRC will occur by the 4th decade of life unless prophylactic colorectal surgery is performed. Extracolonic diseases (principally duodenal and desmoid disease) are now responsible for the persistent gap in life expectancy.2
Diagnosis
Genetic testing will identify a mutation in the APC gene in 85% of cases. In patients where a causative gene cannot be identified then patients should also be tested for other adenomatous polyposis syndromes such as MAP, polymerase proofreading-associated polyposis (PPAP), NTHL1 associated polyposis (NAP) and MSH3-associated polyposis (MSH3AP) (see below). When no identifiable mutation is found then identifying over 100 polyps in the colorectum is pathognomonic for an adenomatous syndrome.
Screening
“At-risk” relatives (ie, first-degree relatives of a patient with a known mutation or with a phenotypic diagnosis) should commence screening from 12 to 14 years . If the gene mutation is known then predictive genetic testing should be offered with appropriate counselling. If the family mutation is identified in the adolescent, the teenager should commence colonoscopy surveillance at age 12–14 years. If the family specific mutation is absent, then the patient can be discharged with no increased lifetime risk of developing CRC. If the genetic mutation within the family is not known then colonoscopy is recommended to start at age 12–14 years until polyps are identified, or repeated 5-yearly until the patient can be entered into a national bowel cancer screening programme (see figure 1).

Familial adenomatous polyposis management flowchart. OGD, gastroduodenoscopy.
Surveillance
Lower gastrointestinal tract surveillance
Colonoscopic surveillance is advised every 1–3 years with intervals determined by the polyp burden. Patients should be referred for colectomy at a convenient time around education and employment for example, age 15–20 years, taking into account colonic adenoma burden. Current practice is to undergo colectomy when there are hundreds of adenomas at 2mm or more, or adenomas >10 mm, or earlier if there is high grade dysplasia.
Upper gastrointestinal tract surveillance
Upper gastrointestinal (UGI) tract surveillance has been recommended, starting from 25 years of age and has been shown to be beneficial.3 Although the data are less, gastric adenomas and cancer are being noticed as newly emerging clinical problems4–6; this may be partly due to increased endoscopic detection. More than 90% of patients will develop duodenal adenomas during their lifetime.3 Duodenal disease can be staged using the Spigelman staging system, which has been validated in terms of predicting 10-year cancer risk. The ampulla is not easily incorporated into the Spigleman staging system and a classification has been proposed by Kashiwagi et al, which does seem to be helpful in terms of determining cancer risk.7 How best to manage the UGI tract is not established. Surveillance does need to provide assessment of the stomach as well as duodenum and ampulla. Surveillance interval is adjusted according to the findings in these areas (see tables 1 and 2). Endoscopic therapy and pancreatic duodenctomy may be considered but there are no current agreed detailed guidelines for UGI tract disease and those with more advanced disease are best referred to a specialist unit.
Classification of the severity of duodenal polyposis as per Spigelman score35
Interval of upper gastrointestinal surveillance
Definitive management of the large bowel
prophylactic surgical management
The primary aim of prophylactic surgery is to prevent CRC thereby removing the colon or colorectum before any adenomas have the chance to progress into cancers.
There are three main surgical options8:
Total colectomy and ileorectal anastomosis (IRA); removing the colon and joining the ileum to the rectum.
Restorative proctocolectomy (RPC/pouch); removing colon and rectum and forming an ileal reservoir thus preserving continence.
Panproctocolectomy; removing colon, rectum and anus and forming an end ileostomy.
Because the latter is often unacceptable to patients the decision in most cases lies between IRA and RPC.
For young patients the advantages of IRA over pouch surgery include better bowel function (there are fewer reports of leakage and less stool frequency compared with pouches), reduced risk of erectile dysfunction secondary to pelvic nerve damage in males (there is a 1%–2% risk with proctocolectomy) and preservation of fertility in females (in RPC pelvic dissection causes fertility to be halved).8
The primary aim of colectomy must be to prevent cancer and there are some patients who are at high risk of disease affecting their rectum, thus RPC is often preferable for patients with more than 500 colonic adenomas, more than 20 rectal adenomas or a rectal adenoma too large for endoscopic removal and a genotype associated with a greater adenoma burden that is, APC mutation 1251 to 1309 9.
Surveillance after prophylactic surgery
Patients who have undergone IRA or RPC require long-term surveillance of 6–12 months. The aim is to manage rectal, pouch body and pouch cuff adenomas endoscopically but surgery may be required if the polyp burden becomes unmanageable for example, secondary proctectomy or pouch excision.
Extracolonic disease
The two major extracolonic manifestations which cause the greatest mortality and morbidity after prophylactic surgery has been performed are duodenal disease (discussed above) and desmoid disease.
Desmoid disease
Desmoid tumours (DT) are non-metastasizing neoplastic tumours that arise from myofibroblasts and occur in different types of connective tissues. Problems arise due to invasion and compression of local structures. 10% to 20% of patients with FAP will develop desmoid disease.10 11 The most common anatomical sites are the small bowel mesentery and abdominal wall. The majority of desmoid lesions remain stable or undergo periods of growth and regression, 10% resolve entirely but 10% demonstrate unabating growth.12 Thus, treatment of desmoid disease is really reserved for lesions that are rapidly growing or causing symptoms.
Medical treatment of desmoid
There are no randomised controlled drug trials for the medical treatment of desmoid disease. Non-steroidal anti-inflammatories (typically sulindac) and antiestrogens (eg, toremifene and roloxifene) are often used although the data supporting their use are weak. Cytotoxic chemotherapy may be considered and the results from case series are promising.
Surgical treatment
Excision of abdominal wall desmoids is generally safe but has a high level of recurrence. Excision of intra-abdominal DT has been reported to be very hazardous, but in a specialist centre with careful case selection outcomes may be acceptable. Decision- making in the management of desmoids is very difficult and referral to a unit experienced in the management of DT is recommended.
MUTYH-associated adenomatous polyposis
MAP is an autosomal recessive condition that is due to bialleic mutations in MUTYH located on chromosome 1. Carrier frequency in the population is reported to be 1:100. Affected individuals can be either homozygotes (location of mutation in the MUTYH gene is identical in both alleles) or compound heterozygotes (each allele carries a different MUTYH mutation). The condition broadly overlaps that of FAP but tends to present with a milder phenotype and a later age of CRC development. Surgical decision-making in terms of managing the colorectum are based on phenotype as described for FAP.
Screening
Genetic testing should be offered to the spouse to determine future risk to their children. If partners are a carrier of the mutation then children should be called for genetic counselling in their late teens. Genetic screening should be offered to all other first-degree relatives.
Surveillance and surgical management
The management of the large bowel in MAP is the same as FAP and based on the polyp burden of the colon and rectum. The UGI management is also the same as FAP but begins 10 years later (there are no reports of gastric disease in MAP).
Endoscopic surveillance of the rectum and pouch will be required postoperatively. If patients have been diagnosed after a diagnosis of cancer and having undergone a segmental resection annual surveillance colonoscopy is recommended for the remaining colorectum and if indicated completion colectomy/ proctocolectomy may need to be performed.
Other adenomatous polyposis syndromes
Improvement in genomic technologies has led to the discovery of newer genes causing adenomatous polyposis such as:
NAP
Constitutional mismatch repair disorder (CMMR-D)
PPAP
MSH3AP
The natural history and tailored surveillance of these subsets of adenomatous polyposis syndromes is yet to be determined.
Serrated polyposis syndrome
SPS is characterised by the occurrence of multiple serrated polyps throughout the colon and the rectum. Prevalence of SPS in the general population is estimated to be 1:2000.13 Previously known as hyperplastic polyposis syndrome, serrated lesions encompass the following terms: hyperplastic polyps, sessile serrated polyps or traditional serrated adenoma.
Diagnosis
The diagnosis of SPS is clinical and based on 2010 WHO definition which are currently undergoing revision. The criteria are:
≥5 serrated polyps proximal to the sigmoid colon; 2 of these must be >10 mm in diameter, OR
Any number of serrated polyps proximal to the sigmoid colon +first-degree relative with serrated polyposis OR
>20 serrated polyps (any size) throughout the colon
Cancer risk and screening
The risk of CRC in SPS is not well understood. Most patients in this group who develop CRC were not aware of their SPS diagnosis until later.
However, those who undergo dedicated surveillance have <2% CRC incidence rate.14 15 There is no known causative genetic mutation to allow predictive testing. All patients should be referred to a clinical genetics unit or a polyposis registry because SPS can overlap genetically and phenotypically with other polyposis syndromes.16 In addition, this allows for protocolised surveillance and appropriate identification and follow-up of family members at risk.
First-degree relatives are advised to commence colonoscopy surveillance from age 25. If serrated polyps are present then 1–2-yearly surveillance colonoscopy will be required; if there are no polyps present then 5 years of surveillance is required. Screening protocols are also currently under revision and with likely WHO criteria-2 changes, a solitary serrated lesion will no longer require 1–2-yearly colonoscopy. In addition, new guidelines may suggest a later commencement of screening or commence 10 years earlier than onset of disease in the index case.
Surveillance
Endoscopic
Careful colonoscopy by an experienced endoscopist is essential as lesions can be difficult to detect and can progress rapidly. Minimum withdrawal time of 6 min and chromoendoscopy may improve detection of serrated lesions.16 Removal of all lesions >5 mm is recommended and once achieved 1–2-yearly surveillance is recommended. There is little evidence to suggest extracolonic cancers. UGI surveillance or extra-luminal surveillance for non-GI cancers are not recommended.16
Surgical management
Surgery is required in approximately 30% of cases.14 17 Surgery should be considered in patients who have lesions that cannot be managed endoscopically due to size, site or number.16 The aim of surgery is to remove all lesions that cannot be endoscopically managed. Options include segmental resection or total colectomy with ileorectal anastomosis.16 The occurrence of metachronous cancers in serrated polyposis is less than previously thought18 and this is likely to impact surgical decision-making in the future.14
Hamartomatous polyposis syndromes
Hamartomatous polyposis syndromes are all rare and their optimal management is still being determined.
Peutz-Jeghers syndrome
PJS is characterised by multiple polyps in the GI tract associated with a mucocutaneous pigmentation with a prevalence of 1:50 000 to 1:200 000.
Diagnosis
A diagnosis can be clinical or genetic. If the genetic mutation is not identified then clinically a patient can be diagnosed with PJS if any one of the following is present19:
Any number of PJ polyps plus family history of PJS.
Characteristic mucocutaneous pigmentations plus family history of PJS.
Two or more PJ polyps which have been confirmed histologically.
Any number of PJ polyps and characteristic mucocutaneous pigmentation.
Presentation
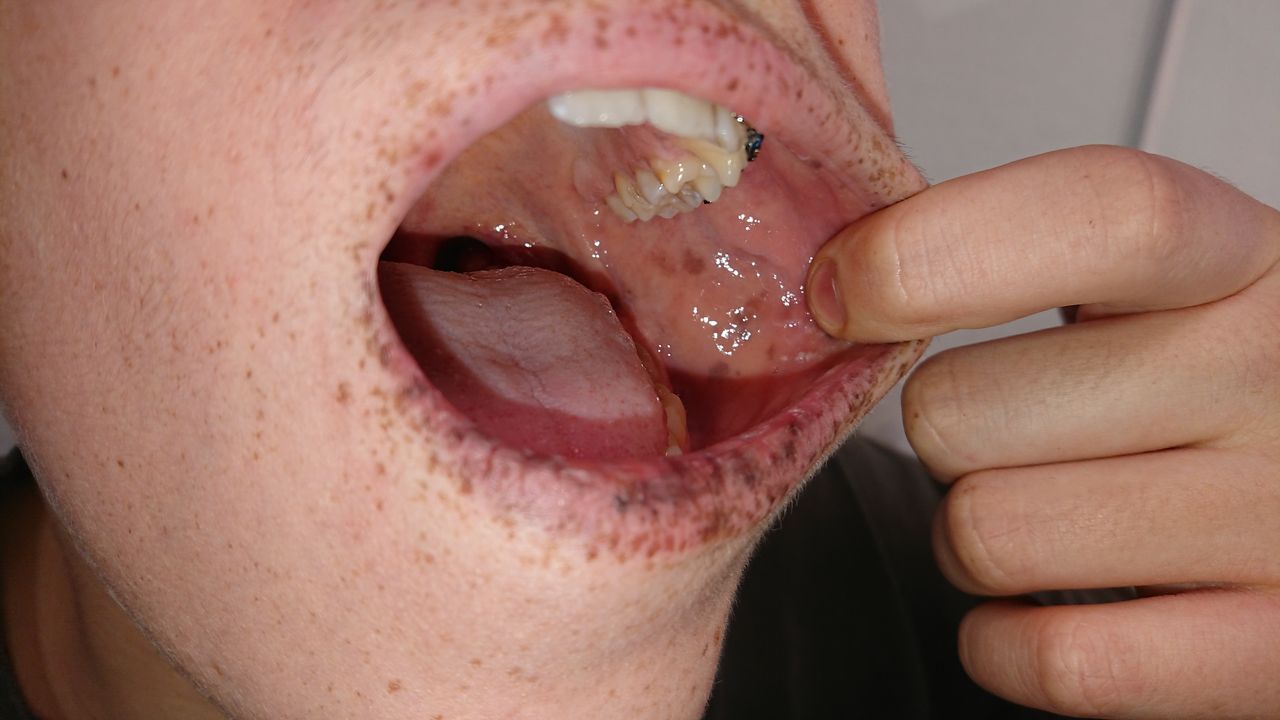
Pigmentation
The mucocutaneous pigmented lesions usually arise in infancy; typical locations are the buccal mucosa, lips, nostrils, perianal area, fingers and toes and dorsal/ volar aspect of hands and feet.19 20 These lesions may fade after puberty (see figure 2).

Picture of Peutz-Jeghers syndrome mucocutaneous pigmentation.
Gastrointestinal
PJS polyps can be located throughout the GI tract but most commonly occur in the small intestine.21 22 Patients can present with intestinal obstruction, intussusception, abdominal pain, per rectal bleeding or prolapsing anal polyps.20 Symptoms from polyps begin in childhood and 33% will have symptoms by the age of 10 and 50% by 20 years of age.19 Prior to the introduction of small bowel surveillance, 70% of PJS patients developed intussusception before the age of 18 years, for many patients necessitating emergency laparotomy with bowel resection. Extra-intestinal polyp sites have also been described but are rare.23
Cancer risk
Although the cancer risk in PJS is likely to be overestimated due to ascertainment bias, it is accepted that patients have a significantly raised cancer risk to that of the general population of a number of cancers (see table 3). Historically GI malignancies were thought to be the most common but more recently breast and pancreatic cancers were the most common cancers arising in a cohort on surveillance.
Cumulative cancer risk in Peutz-Jeghers syndrome36
Screening
The condition is due to a mutation in the STK11 (also known as LKB1) gene located on chromosome 19q13.3.24 25 Genetic testing should be offered to at-risk relatives from age 3. Screening endoscopy is recommended for families without a known germline mutation.
Management
Of all the syndromes, PJS is the polyposis condition that requires the most clinical attention during childhood. Management is focused on identifying small bowel polyps and managing polyp-related complications (eg, intussusception and bleeding).
Surveillance
Gastrointestinal
GI surveillance should commence no later than at age 8 years, or earlier if symptomatic for example, unexplained abdominal pain,+/-vomiting. Affected children and teenagers should undergo gastroscopy and placement of video capsule endoscopy (VCE) every 3 years to visualise small bowel polyps or MR enterography depending on local expertise (figure 3). Small bowel polyps measuring >2 cm (or smaller if symptomatic) should be removed by balloon-assisted enteroscopy, or laparoscopy or laparotomy with intraoperative enteroscopy to clear the small bowel of polyps. Colonoscopic surveillance should commence at age 8, or earlier if there is rectal bleeding. Once polyps have been identified in colonoscopy, this should be repeated 3-yearly, but if no polyps are identified at colonoscopy, then the next colonoscopy can be delayed until age 18 years, unless there is rectal bleeding.
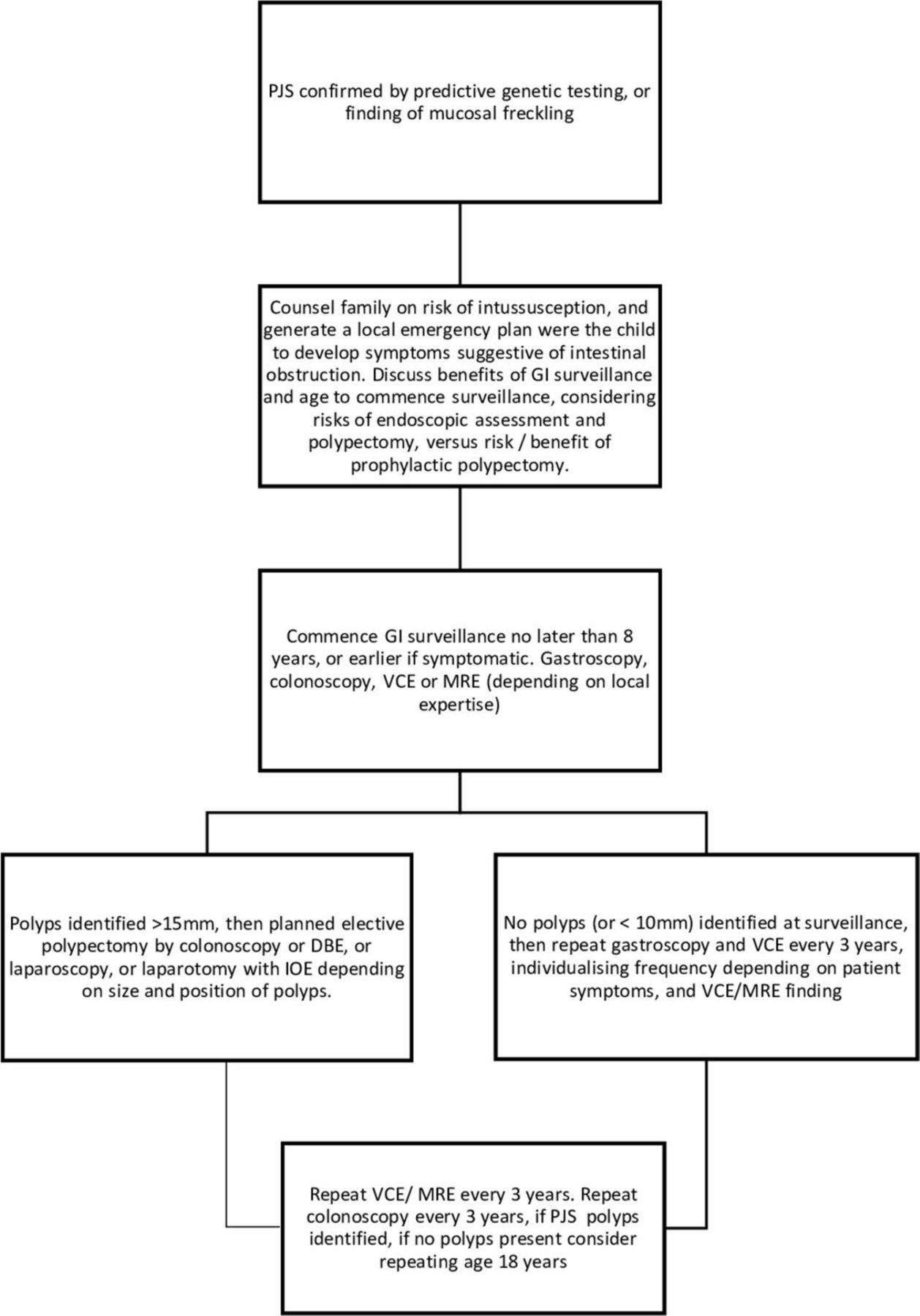
{kind=link}
{kind=link}
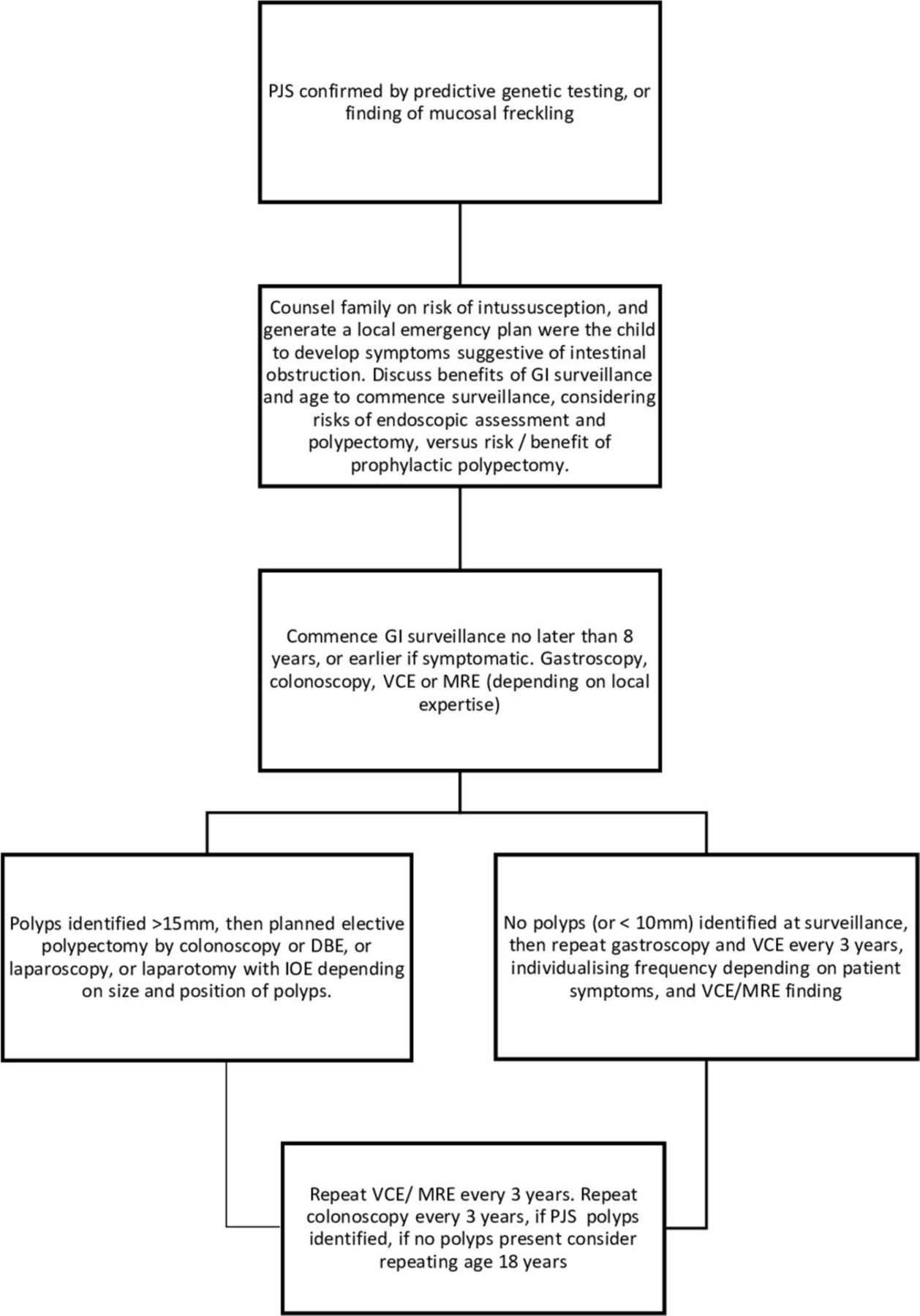{kind=link}
GI surveillance in PJS syndrome. BDE, balloon-assisted enteroscopy; GI, gastrointestinal; IOE, intraoperative enteroscopy; MRE, MR enterography; PJS, Peutz-Jeghers syndrome; VCE, video capsule endoscopy.
Breast and cervical screening
Females should undergo annual mammogram from 30 years of age and if there is a family history of breast cancer patients should be referred for consideration of genetic testing (BRCA). Females should have cervical smear as per national screening programme.
Managing the symptomatic patient with intussuception
Symptomatic patients presenting with small bowel obstruction secondary to intussusception should be referred urgently for surgery, usually laparotomy to remove small bowel polyps >2 cm or for any sized polyps causing abdominal pain; the entire length of small bowel should be inspected with intraoperative enteroscopy at this opportunity to avoid multiple re-laparotomies.
Juvenile polyposis syndrome
JPS has an incidence of 1 :100 000.26 ‘Juvenile’ refers to histopathological features of the polyps rather than age of onset. Polyps are found predominantly in the colon, particularly on the right side, but are also seen in the UGI tract. Patients are at an increased risk of cancer. The cumulative life-time risk of CRC is estimated to be 30%27 and in addition there is an increased risk of gastroduodenal cancer.28 29
Extra-intestinal clinical problems include cardiovascular (eg, mitral valve prolapse, telangiectasia, arterio-venous malformations and aneurysms) and neurological malformations (eg, macrocephaly, hydrocephalus and cleft palate).30
Diagnosis
A diagnosis of JPS can be made by meeting any one of the following criteria31:
>5 juvenile polyps of the colorectum.
Juvenile polyps throughout the GI tract
Any number of juvenile polyps with a family history of juvenile polyposis
One severe phenotype of JPS is juvenile polyposis of infancy, presenting before age-1 with multiple GI polyps, anaemia and hypoalbuminaemia.
Presentation
The majority of patients present symptomatically with rectal bleeding, prolapsing polyp, melaena, abdominal pain and diarrhoea32 with a median age of presentation of 27 years.30
Screening
Genetics
JPS has an autosomal dominant pattern of inheritance and thus far mutations in two genes have been found; SMAD4 and BMPR1A. SMAD4 mutations are associated with higher frequency of UGI polyps and may overlap with hereditary haemorrhagic telangiectasia (HHT). Rarely patients can have adjoining deletions of both BMPR1A and PTEN which lie close to each other on chromosome 10q22-23. These patients seem to have a more severe JPS phenotype combining some features of Bannayan-Riley-Ruvalcaba syndrome (BRRS) and JP of infancy.33
In families where the gene mutation has been detected predictive genetic testing should be offered to at-risk children from the age of 12–14 years.
JPS surveillance
GI surveillance
Surveillance of the GI tract is recommended. Colonoscopy should start at the age of 15 (sooner if symptomatic) and then repeated at a 1–3-yearly interval.30 UGI endoscopy is recommended to start at age of 18, although with better data, genotype specific recommendations may be required.
Extra-intestinal surveillance
Patients should have an annual cardiovascular examination. Patients with a SMAD4 mutation should be assessed for features of HHT. This involves investigating with ECG, CT chest and VCE34
JPS surgery
Most cases can be managed with therapeutic endoscopy with polypectomy but surgery is required for patients who develop cancer or where endoscopic management is not possible.
PTEN hamartoma tumour syndrome
PTEN hamartoma tumour syndrome is an umbrella term for various clinical syndromes caused by a germline mutation of the PTEN tumour suppressor gene. It includes Cowden syndrome, BRRS, PTEN-related Proteus syndrome and Proteus-like syndrome. These are extremely rare conditions that require referral to a local genetics centre.
Family planning
Previously, patients who were aware of their diagnosis and wanted to have children were restricted in terms of their family planning to either choosing not to have biological children of their own or conceiving and having a 50% chance of passing on the mutated gene to their offspring. Prenatal testing such as amniocentesis or chorionic villus sampling were unacceptable to some patients due to the stage of the pregnancy at which they are performed. Currently however, where the family mutation is known (except in serrated polyposis) patients can undergo in vitro fertilisation with preimplantation genetic diagnosis (PGD). Only embryos that have a normal copy of the gene are then placed back into the uterus.
Summary
Polyposis syndromes are complex in presentation, diagnosis and management. Where possible patients should be referred to a genetics centre or a polyposis registry so that protocolised management can be instigated and dedicated follow-up of patients and ‘call up’ of at-risk relatives can be implemented.
References
Footnotes
Contributors RP and WH jointly wrote the manuscript. RP is funded by Bowel Cancer UK - Royal College of Surgeons one year research fellowship.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Obtained.
Provenance and peer review Commissioned; externally peer reviewed.
Linked Articles
- UpFront