Article Text

Abstract
Background and aim MUTYH-associated polyposis (MAP) is an autosomal recessive inherited disorder characterised by the development of polyposis in the upper and lower gastrointestinal tract and a high risk of colorectal cancer (CRC). We evaluated the natural history of the disease and the outcome of colorectal surveillance and management.
Methods A large Western European dataset of biallelic MUTYH mutation carriers comprising 254 patients was used. Detailed information was collected on polyp and cancer development in the colorectum, and the outcome of surveillance and surgery. Survival methods were used to calculate the risk of CRC development.
Results The mean follow-up was 9.8 years. Colorectal polyposis was diagnosed at a mean age of 44.8 years (range: 12–77 years). Most patients had <100 colorectal adenomas at diagnosis. CRC was diagnosed in 147 (58%) of the 254 patients (mean age at diagnosis: 48.5, range: 21–77 years). The cumulative lifetime risk of CRC was 63% at age 60 years. There was no correlation between the number of adenomas and the presence of CRC. The cumulative risk of CRC in patients presenting with polyps was 9% after 5 years of follow-up. Patients presenting with CRC had 11% risk of developing a metachronous CRC at 5 years after surgery. Thirty-seven per cent of patients with MAP with CRC who underwent partial colonic resection needed secondary surgery shortly afterwards.
Conclusions The high risk of developing CRC under surveillance in patients with MAP may suggest an accelerated carcinogenesis. Surveillance of these patients should therefore include colonoscopy at short intervals, for example, at 1–2-year intervals starting from the age of 18 to 20 years. If surgery for CRC is warranted, a (sub)total colectomy is recommended.
- MUTYH-associated polyposis
- MAP
- colorectal cancer
- polyposis
Statistics from Altmetric.com
Significance of this study
What is already known about this subject?
MUTYH-associated polyposis (MAP) is a recessive inherited polyposis syndrome caused by mutations in the base-excision repair gene MUTYH
The MAP phenotype resembles that of attenuated familial adenomatous polyposis
The MAP extracolonic tumour spectrum resembles that of Lynch syndrome
What are the new findings?
In patients with MAP, colorectal cancer (CRC) risk is not associated with the number of colorectal polyps
CRC development seems to be accelerated, as about 10% of the patients presenting with polyposis or CRC had developed a primary or a metachronous CRC within 5 years of follow-up
After hemicolectomy, patients had a substantial risk of reoperation
How might it impact on clinical practice in the foreseeable future?
Biallelic MUTYH mutation carriers should have regular colonoscopic surveillance independent of the number of polyps. If surgery is needed, a total colectomy is recommended
Introduction
The best known inherited form of gastrointestinal polyposis is familial adenomatous polyposis (FAP), an autosomal dominant syndrome caused by APC germline mutations. MAP is an inherited polyposis syndrome described for the first time in 2002 that is transmitted as an autosomal trait caused by biallelic germline mutations in the base-excision repair gene MUTYH (OMIM #608456), located on chromosome 1. The syndrome is characterised by the development of multiple colorectal adenomas and a high risk of CRC.1 The frequency of monoallelic mutations in the Western European population is about 2%. Approximately, 0.3% of patients with CRC from population-based series are associated with biallelic MUTYH mutations.2
Usually, biallelic MUTYH mutation carriers present with a polyposis phenotype that strongly resembles that of AFAP, with onset in the fourth or fifth decade, and development of <100 colorectal polyps predominantly in the right colon.3 Duodenal adenomas and carcinomas are reported in patients with FAP and those with MAP.4 5 A recent study indicated that the overall incidence of extraintestinal malignancies was increased. The reported tumour spectrum included cancer of the ovary, bladder and skin, which indicates an overlap with the Lynch syndrome.4
The risk of developing CRC is comparable to that in FAP, although the age at onset is delayed.3 A population-based series showed high penetrance of biallelic mutations with a substantially elevated CRC risk, with estimated penetrances of 20% at age 50 and 43% at age 60 years, respectively.2 Compared to the general population, relative risks of CRC were estimated from 53 to 117.6 7 For monoallelic mutation carriers, a trend of a slightly elevated CRC risk was suggested in most studies.6 8 9
Currently, limited information is available on the natural history of adenoma and carcinoma development in MAP. The outcome of surveillance and surgical management is also unknown. The question is whether MAP is simply an attenuated version of FAP or a distinct cancer susceptibility syndrome needing specific management guidelines.10 11
The aims of the present study were to evaluate the natural history of adenoma and carcinoma development and to assess the outcome of management of a large series of patients with MAP.
Methods
Patients
For this study, clinical data were retrieved from three genetics institutes (Institute of Human Genetics, Bonn, Germany; Institute of Medical Genetics, Cardiff, UK; Centre for Human and Clinical Genetics, Leiden, The Netherlands) and The Netherlands Foundation for the Detection of Hereditary Tumours. Ethical approval was obtained from national and/or local review boards (The Multi-Centre Research Ethics Committee for Wales, ref. 06/MRE09/19; medical faculty of the University of Bonn Ethics Review Board, no. 063/04; Leiden University Medical Centre Ethics Review Board, no. P01.019). The methods for MUTYH mutation analysis in patients with adenomatous polyposis have been described previously.3 12 13
Patients with biallelic MUTYH mutations with available data on management and follow-up were selected. The study cohort consisted of patients with symptoms (75%) and call-up cases (25%) and overlaps with the cohort described in previous studies.4 9 14
Data and statistical analysis
The data collected include gender, mutation, date of birth and details on diagnosis, disease course of polyposis and CRC development. Available data on surgery were analysed. Data are presented as mean values (with ranges) for continuous variables and numbers (with percentages) for categorical variables. To calculate the risk of cancer development and the probability of secondary surgery, Kaplan–Meier methods were used. The observation time was from age at diagnosis of polyposis to event, death, loss to follow-up or end of the study (1 July 2009). Data were analysed and calculated with SPSS V.16.0.0 (SPSS Inc.). A p value of <0.05 was considered to be statistically significant.
Results
Characteristics of the study cohort
Data were available on 254 biallelic MUTYH mutation carriers, of whom 141 (56%) were male. They presented between 1963 and 2009 with polyposis and/or a CRC, or were identified due to mutation analysis in the family. The mean duration of follow-up from diagnosis of polyposis to the last observation for this study was 9.8 years (range: 10 months–36 years). The mean age at diagnosis of polyposis was 44.8 years (range: 12–77 years). The majority of patients (63%) had <100 colorectal polyps at first diagnosis; 20% had more than 100 adenomas, and in 15% of patients, the exact number of adenomas was unknown. Four patients (2%) had no polyps at the time of diagnosis; two of them presented with CRC, and two underwent colonoscopy due to positive mutation analysis in the family.
CRC development
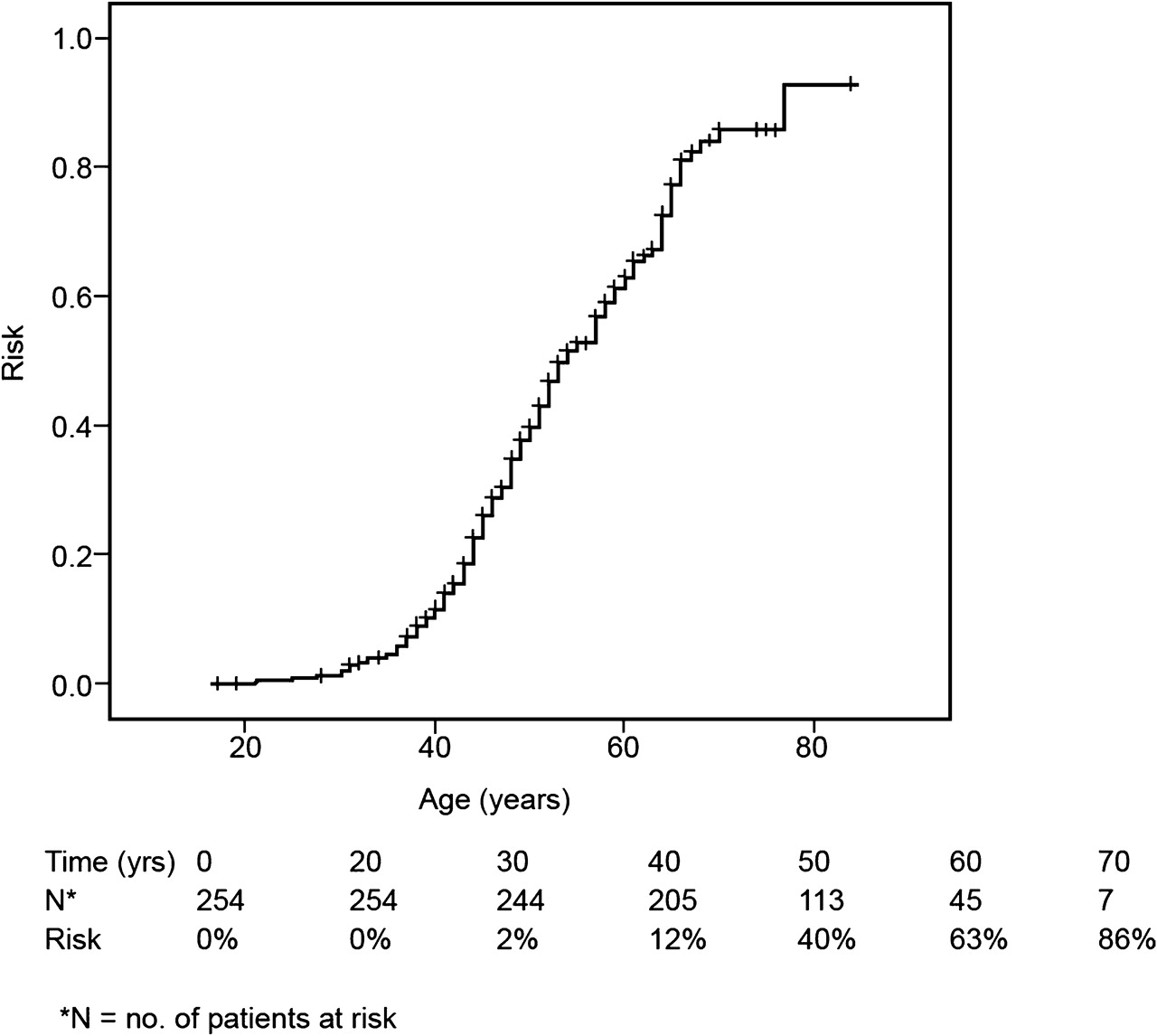
A total of 147 (58%) of the 254 patients developed CRC at a mean age of 48.5 years (range: 21–77 years). Three patients (2%) were younger than 30 years at diagnosis of CRC. The lifetime cumulative risk of developing CRC is shown in figure 1. By the age of 60 years, 63% had developed CRC. Eighty (54%) of the 147 CRCs were right sided, whereas 56 (38%) were located in the left part of colon and rectum; for 11 CRCs, the location was unknown.

Cumulative risk of developing CRC for biallelic MUTYH mutation carriers. CRC, colorectal cancer.
In 120 patients, CRC was diagnosed at the initial endoscopy at a mean age of 48 years (range: 21–77 years). Table 1 displays the number of colorectal polyps detected simultaneously with CRC.
Number of polyps at initial endoscopic examination in patients presenting with CRC (n=120)
About half of the patients had <50 adenomas. The risk of developing CRC was comparable for patients with <50 and those with >50 colorectal polyps (43% vs 46%, p=0.647). Histopathology analysis showed adenomas in 118 patients. Two patients who presented with CRC had no colorectal polyps. In 12 patients (10%), besides adenomas, hyperplastic polyps were also reported; in all cases, the majority of polyps was adenomatous.
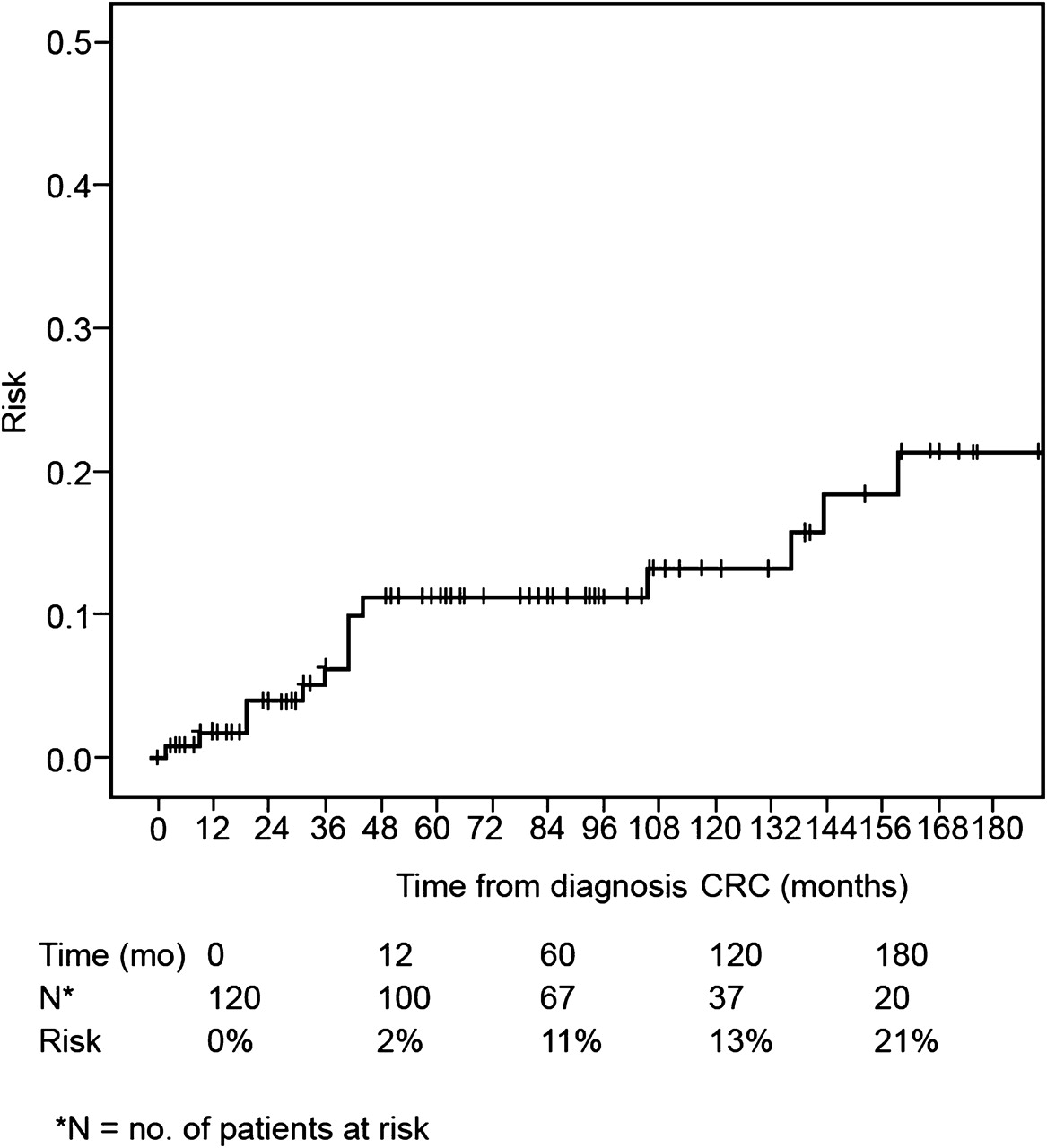
The remaining group of 134 patients presented with ‘polyposis only’ at a mean age of 42 years (range: 12–68 years). The initial treatment of these patients was endoscopy and polypectomy in 70, surgery in 34 and unknown in 30 patients. Twenty-seven (20%) of these patients subsequently developed CRC during follow-up at a mean age of 52 years (range: 36–68 years). The cumulative risk of developing CRC during follow-up is shown in figure 2. Within the first year after diagnosis of polyposis, the risk of developing CRC was 5%, increasing to 9% after 5 years and to 31% after 15 years of follow-up. In addition, 16 of the 120 (13%) patients who presented with primary CRC developed a metachronous CRC under surveillance. The cumulative risk of developing a metachronous CRC is shown in figure 3. In the first year after primary CRC, 2% of the patients had developed a metachronous tumour, increasing to 11% after 5 years and to more than 20% after 15 years. Features of patients with primary CRC and those with CRC detected under surveillance are shown in table 2.

Cumulative risk of developing CRC under surveillance after presenting with polyposis. CRC, colorectal cancer.
{kind=link}
{kind=link}
{kind=link}
Cumulative risk of developing metachronous CRC in patients presenting with CRC. CRC, colorectal cancer.
Characteristics of CRC in patients with biallelic MUTYH mutations, according to mode of diagnosis
Surgical management
Detailed information on surgical management was available for 87 of 120 patients with CRC at initial endoscopy. Fourteen (37%) of 38 patients who had had a partial colectomy needed reoperation, including seven because of cancer and seven because of uncontrollable polyps that could not be removed endoscopically. Only four (8%) of 49 patients undergoing a (sub)total colectomy needed reoperation, all because of polyps.
Discussion
Previous studies have shown that the phenotypic expression of MAP resembles that of AFAP. The majority of patients with MAP and those with AFAP develop <100 colorectal adenomas and present with CRC at a more advanced age compared to those with classical FAP. However, the present study demonstrates that there are also remarkable differences between the two polyposis syndromes. In MAP, in contrast with (A)FAP, the risk of CRC appears not to be associated with the number of adenomas in the colorectum. Another clinically important finding was that a substantial proportion of patients developed CRC within the first decade after primary diagnosis of polyposis or a primary CRC, an observation that suggests an accelerated adenoma-to-carcinoma progression. Regarding the surgical treatment of CRC, more than one third of the patients who underwent partial resection needed secondary surgery, often soon after primary treatment.
In classical FAP caused by an APC mutation, there is a strong correlation between the polyp burden and CRC risk. For example, patients with the codon 1309 mutation generally develop thousands of colorectal adenomas in the second decade of life, and these patients have a high risk of developing CRC before the age of 30 years.15 In contrast, patients with attenuated FAP develop less adenomas and develop CRC on average in their 50s. A recent study on patients with AFAP showed that the median number of colorectal adenomas in patients with CRC was higher than that in patients without CRC (37 vs 20, p=0.05), suggesting that, also in the AFAP group, the cancer risk is related to the number of adenomas.16 In our series of patients with MAP, the risk of CRC was not clearly associated with the number of adenomas. About 40% of the patients with CRC had <50 adenomas, and a similar proportion had >50 adenomas. Two patients even developed CRC in the absence of adenomas. A phenotype without polyps has also been observed in biallelic MUTYH mutation carriers identified in population-based CRC series. A substantial proportion (29%) of population-based patients with CRC with biallelic MUTYH mutations did not have adenomas.2 17 These findings have important implications for clinical practice because they suggest that the polyp burden may not be a good guide for determining the surveillance interval in patients with MAP and that biallelic mutation carriers without colonic adenomas should also have colonoscopic examinations at short intervals. Furthermore, the absence of polyps does not exclude the diagnosis of a hereditary polyposis syndrome.
The most remarkable finding in our study was the high risk of CRC development in patients under surveillance. In patients presenting with polyposis as well as in patients presenting with CRC, the risk of developing a primary or metachronous CRC within 5 years of follow-up was considerable (9% and 11%, respectively). This risk is even higher than the risk of developing CRC under surveillance reported for Lynch syndrome, which was 6% at 10 years of follow-up.18 Along with our observation of an advanced stage (regional metastases) in one third of the screen-detected CRCs, these findings may indicate the presence of an accelerated carcinogenesis in MAP that has also been reported for Lynch syndrome.19 There are parallels between MAP and Lynch syndrome as defects in DNA repair function are involved in both syndromes, that is, base excision repair and DNA mismatch repair, respectively. In Lynch syndrome, the mismatch repair defect leads to rapid accumulation of mutations in genes that control cell growth and division, which may explain the accelerated carcinogenesis. The mechanism for an accelerated carcinogenesis in MAP is unclear. As suggested in recent studies, there may be a link between base excision repair and low-frequency MSI pathways.17 Recently, it was suggested that the MLH1 gene can be a target of MUTYH transversions leading to MSI phenotype.20 Another study showed some similarities of MAP-associated CRCs to microsatellite unstable cancers, such as a preferential right-sided location, mucinous histology type and increased presence of tumour-infiltrating lymphocytes.21 Furthermore, as adenomas and hyperplastic polyps were found in patients with MAP, the serrated pathway is thought also to play a role in MAP tumourigenesis.22
The usual surgical treatment for FAP is a (sub)total colectomy with either an ileorectal anastomosis or an ileo-pouch anal anastomosis. In the present study, a substantial proportion of patients had undergone a hemicolectomy. In view of the high probability of reoperation after hemicolectomy in our series, ileorectal anastomosis seems to be the best surgical option in MAP, including patients with CRC and only a few colorectal adenomas. In patients with multiple adenomas in the rectum, an ileo-pouch anal anastomosis might also be an appropriate option. As CRC is rare before the fourth decade, the optimal timing for prophylactic colectomy may be later than in classic polyposis.23 Colonoscopy at intervals of 1–2 years and polypectomy might be considered in patients with MAP with few adenomas without CRC.
The present study is based upon a large series of biallelic MUTYH mutation carriers identified via three polyposis registries in the UK (Cardiff), Germany (Bonn) and the Netherlands (Leiden). As a large proportion of patients with symptoms were referred to a geneticist, we have to keep in mind the possibility of bias towards cases with symptoms with multiple polyps and an overestimation of the risk of developing CRC in asymptomatic patients. This explains the finding of multiple polyps in 98% in our cohort (table 1) compared to 70% in biallelic mutation carriers identified in population-based CRC series.2 17
In polyposis registries, usually, annual or biannual colonoscopies are recommended in patients with multiple adenomas from polyposis families with either an APC defect or an MUTYH defect and also in families with an unknown gene defect. A limitation of our study is that we were not always informed whether such a protocol was consequently applied in all three registries or that we had no detailed information on management strategies. However, the fact that the risk of developing metachronous CRC after surgical treatment of CRC (after which a strict colonoscopic protocol is advised) was virtually the same as the risk observed in patients who presented with polyposis suggests that accelerated carcinogenesis is a true feature of MAP. Future prospective studies are needed to confirm our findings.
Another limitation is that we are not certain whether all metachronous lesions were real metachronous lesions and not missed synchronous adenomas or CRCs. Although colonoscopy is the gold standard for colonic examinations, studies have shown that even advanced lesions may remain undetected.24 However, even if some of the lesions were missed lesions, the risk of developing CRC is still considerable.
In conclusion, the recently recognised syndrome of MAP appears to be a cancer susceptibility syndrome with a distinct phenotype with features associated with FAP and Lynch syndrome. MAP resembles (attenuated) FAP phenotypically with respect to numbers of adenomas and age at diagnosis of CRC. However, as shown recently, the tumour spectrum overlaps that of Lynch syndrome, and the accelerated CRC development observed in MAP is also a typical feature of Lynch syndrome. Based on these findings, we propose an intensive colorectal surveillance program for proven biallelic MUTYH mutation carriers consisting of regular colonoscopic screenings every 1–2 years independent of the number of adenomas. In patients with CRC, unmanageable polyps or adenomas with a high degree of dysplasia, a (sub)total colectomy is the most appropriate surgical option.
Acknowledgments
Mallorca Group for helpful discussions.
References
Footnotes
Funding Wales Gene Park, Cardiff, United Kingdom Dutch Digestive Disease Foundation (grant no. MWO 0355), Nieuwegein, the Netherlands.
Competing interests None.
Ethics approval This study was conducted with the approval of The Multi-Centre Research Ethics Committee for Wales, ref. 06/MRE09/19; medical faculty of the University of Bonn Ethics Review Board, no. 063/04 and Leiden University Medical Centre Ethics Review Board, no P01.019.
Provenance and peer review Not commissioned; externally peer reviewed.