Article Text

Abstract
Cholangiopathies describe a group of conditions affecting the intrahepatic and extrahepatic biliary tree. Impairment to bile flow and chronic cholestasis cause biliary inflammation, which leads to more permanent damage such as destruction of the small bile ducts (ductopaenia) and biliary cirrhosis. Most cholangiopathies are progressive and cause end-stage liver disease unless the physical obstruction to biliary flow can be reversed. This review considers large-duct cholangiopathies, such as primary sclerosing cholangitis, ischaemic cholangiopathy, portal biliopathy, recurrent pyogenic cholangitis and Caroli disease.
- primary sclerosing cholangitis
- biliary cirrhosis
- biliary strictures
- portal hypertension
- endoscopic retrograde pancreatography
Statistics from Altmetric.com
- primary sclerosing cholangitis
- biliary cirrhosis
- biliary strictures
- portal hypertension
- endoscopic retrograde pancreatography
Introduction
‘Cholangiopathy’ refers to a disorder of the biliary system and describes a group of conditions affecting the intrahepatic and extrahepatic biliary tree. Impairment to bile flow and chronic cholestasis cause biliary inflammation and provoke ductular and periportal fibrosis, which lead to more permanent damage such as destruction of the small bile ducts (ductopaenia) and biliary cirrhosis. Most cholangiopathies are progressive and cause end-stage liver disease unless the physical obstruction to biliary flow can be reversed.1 This review considers large-duct cholangiopathies, such as primary sclerosing cholangitis (PSC), ischaemic cholangiopathy, portal biliopathy, recurrent pyogenic cholangitis and Caroli disease, focusing on clinical management and intervention including endoscopic biliary therapy.
Pathophysiology of cholangiopathy
Cholangiocytes are specialised biliary epithelial cells lining the biliary tree which modify the volume and composition of bile by regulating the movement of bile, water, electrolytes and solutes across the biliary epithelium.2 3 They can be activated by endogenous and external stimuli and play a critical role in liver injury and repair.4 When cholangiocytes are challenged by stimuli in disease states, they release proinflammatory cytokines which modulate cholangiocyte proliferation and apoptosis, with downstream consequences such as fibrosis, angiogenesis and recruitment of mesenchymal cells.1 5 6 Although acute injury is often reversible, iterative processes of chronic inflammation and fibrosis eventually lead to irreversible damage and duct destruction (ductopaenia), periportal fibrosis and biliary cirrhosis.6 All cholangiopathies have commonality in the cholangiocentric inflammatory processes that lead to the development of fixed and ultimately irreversible biliary injury.1 7
Primary sclerosing cholangitis
PSC is characterised by inflammation, fibrosis and multifocal stricturing of medium/large ducts in the intrahepatic and extrahepatic biliary tree, leading to complications of cholestasis and ultimately liver failure.8 The pathogenic mechanisms in PSC are probably a composite of autoimmune injury compounded by genetic and environmental factors that subvert normal immune surveillance and trafficking, altered gut permeability and impaired innate immune responses. Unlike primary biliary cholangitis, bile salt therapies such as ursodeoxycholic acid (UDCA) have not been convincingly demonstrated to be of benefit in PSC,9 although this may just reflect the fact that manipulation of bile salts is largely ineffective in advanced disease where cholestasis is physically determined by large/medium bile duct stricturing and occlusion. Data on any form of effective therapy are currently lacking, although recent research has highlighted the importance of interactions between gut microbiota and host immune responses as a key target in the pathophysiology of PSC and bile homeostasis.
Epidemiology
The typical presentation of PSC occurs in men (65%) aged 30–40 years with a history of inflammatory bowel disease (IBD).10 The majority of patients with PSC have IBD, typically a unique form of colitis which commonly exhibits right-sided predominance, ileitis and rectal sparing. The prevalence of PSC in ulcerative colitis (UC) is around 5%.10
PSC can be considered to be a premalignant state with a clear increase in the risk of both biliary and colorectal cancer,11–13 which necessitates routine surveillance of the colon and gall bladder. Rates of colorectal cancer in PSC colitis are increased fourfold when compared with patients with UC alone, with a 1:3 likelihood of developing colorectal cancer over 20 years, often within the right colon.14
Clinical features
Fatigue, pruritus and jaundice can be present in patients at the time of diagnosis, but many patients tend to be asymptomatic, with PSC often detected as part of evaluation of liver function tests, particularly in patients with IBD.15
Investigations
Liver function tests generally demonstrate an elevated alkaline phosphatase (ALP), with both ALP and bilirubin fluctuating in relation to intermittent biliary obstruction due to sludge/stones. Albumin levels tend to be normal but may be low with advanced liver disease or in conjunction with active IBD. Hypergammaglobulinaemia (30%) and/or raised IgM levels (40%–50%) are often present, while positive perinuclear antineutrophilic cytoplasmic antibodies (30%–80%), human leucocyte antigen (HLA-DRw52a) (52%–100%)16 17 and antismooth muscle antibodies and antinuclear antibodies may be seen.
Radiological investigations such as transabdominal ultrasonography and CT may identify bile duct thickening, segmental or main duct dilatation, mass lesions and features of portal hypertension.18 Magnetic resonance cholangiopancreatography (MRCP) is the investigation of choice to demonstrate abnormalities of the biliary tree, such as multifocal stricturing, sacculation and dilatation of the intrahepatic/extrahepatic bile ducts. Endoscopic retrograde cholangiopancreatography (ERCP) should not be considered as an initial diagnostic investigation, but in patients who have contraindications to MRCP (eg, those with implanted metal devices) ERCP may be required.18 19 Small-duct PSC is a variant of PSC where cholangiography is normal, although the biochemical and histological features are similar to classical large-duct PSC: while small-duct PSC has a better prognosis, it may evolve into medium-duct/large-duct disease.20 21
A liver biopsy is rarely diagnostic but may be helpful in small-duct PSC or when an overlap syndrome (PSC associated with autoimmune hepatitis) is suspected. Characteristic histological findings in PSC are fibrous obliteration of the small bile ducts with concentric replacement by connective tissue in an ‘onion skin’ pattern (figure 1), but this is only seen in around 25% of biopsies, as most biopsies demonstrate non-specific features. Biopsies quantify the degree of liver fibrosis, although fibrosis is increasingly assessed through the use of transient elastography with a receiver operating characteristic curve of 0.96, using a cut-off of 17.3 kPa for F4 fibrosis.22

Histological features of primary sclerosing cholangitis (PSC). Characteristic concentric ‘onion-ring’ fibrosis and thickening of a bile ductule (*) is noted. The hepatic artery is indicated by the arrow.
Management
In early-stage PSC (before the onset of fibrosis), standard-dose UDCA may have a therapeutic role, but high-dose UDCA should not be prescribed as trials have demonstrated harm at doses of 28–30 mg/kg/day.23
Mechanical obstruction of biliary segments results in recurrent cholangitis and repeated inflammatory injury to the biliary epithelium. A ‘dominant’ stricture is defined as stenosis of the common bile duct of ≤1.5 mm diameter or ≤1 mm in the intrahepatic ducts14 (figure 2). Patients with PSC have a 10%–15% lifetime risk of developing cholangiocarcinoma, a rate 30 times higher than the general population, as well as an increased risk of gallbladder cancer.24 The highest rates are seen in those with PSC-IBD, where the clinical course is usually aggressive and often precludes liver transplantation. Malignant gallbladder polyps are more common in PSC, and it is reasonable to consider cholecystectomy for gallbladder polyps even if <1 cm in size owing to their high risk of malignant transformation if liver function will permit surgery.25

Typical cholangiographic appearances of the biliary tree in primary sclerosing cholangitis with multifocal stricturing, sacculation and dilatation of the intrahepatic bile ducts (white arrow) and a dominant stricture in the common bile duct (black arrow).
Diagnosing cholangiocarcinoma can be challenging in PSC, and cross-sectional imaging (CT, MRCP) followed by endoscopic ultrasound (EUS) evaluation is favoured in many units, with ERCP-based techniques such as brush cytology/biliary biopsies of dominant strictures and cholangioscopy used in selected cases for direct ductal evaluation and biopsies.26 27 Great caution should be exercised in selecting patients for endoscopic intervention, and cases should be discussed with a hepatobiliary centre before proceeding. Strictures should be balloon-dilated and long-term biliary stents should be avoided due to the risk of precipitating cholangitis.28 29 Patients with PSC and UC have an increased risk of colorectal cancer and require annual colonoscopy screening,30 with transabdominal ultrasound surveillance to identify and monitor gallbladder polyps.14 31
Liver transplantation
Prioritisation for liver transplantation in advanced liver disease is based on symptoms and validated scores such as United Kingdom Model for End-Stage Liver Disease (UKELD), although recent practice has been to consider patients with PSC for transplantation at an earlier stage before refractory cholangitis becomes established. Many patients are listed for symptoms such as recurrent cholangitis. Concurrent IBD may be associated with a worse prognosis and lower transplant-free survival.32 Moreover, the risk of developing pouchitis in PSC-IBD is significant, and the presence of an ileoanal pouch increases the risk of hepatic artery thrombosis and graft dysfunction.
Ischaemic cholangiopathy
Ischaemic cholangiopathy results from impaired arterial perfusion to the bile ducts, which are exclusively supplied by the hepatic artery through the peribiliary plexus.33 Ischaemic cholangiopathy commonly occurs postliver transplantation but can also occur due to vascular injury during biliary surgery, sickle crises, chemoembolisation of liver tumours, major trauma and hypercoagulable states.34 35
Biliary complications of liver transplantation are not uncommon and have been reported to occur in 15%–40% of transplants.36 Strictures following orthotopic liver transplantation can occur at the biliary anastomosis or be more diffuse.34 37–40
Anastomotic strictures
Anastomotic strictures develop in the early postoperative period where they are usually related to technical issues and oedema at the anastomosis, and the vast majority resolve promptly following a period of endoscopic stenting.41–43 Anastomotic strictures occurring more than 6 months after surgery are fibrotic lesions that have a higher likelihood of recurrence after endoscopic intervention. Their aetiology is more complex as they may reflect hepatic arterial insufficiency or peritransplant ischaemia and often present insidiously. If left untreated, these strictures cause cholangitis and biliary cirrhosis.
Non-anastomotic strictures
Non-anastomotic strictures (NAS) reflect a heterogeneous collection of lesions that commonly occur at the hilum and involve first-order and second-order bile ducts.37 Unlike anastomotic strictures, NAS are much more common in organs retrieved from DCD (donation after cardiac death) donors,44 where the risk of ischaemic injury is higher due to the longer periods of normothermic ischaemia, and in ABO-incompatible live donor transplantation where antibody-mediated rejection can cause biliary stricturing. It is hoped that the use of machine-perfused organs will reduce the risk of peritransplant graft ischaemia.
Ischaemic biliary strictures
Ischaemic biliary strictures can occur in up to 7%–9% of DCD grafts.45 46 The characteristic lesion is a complex multifocal perihilar stricture occurring between 1 and 6 months after surgery (figure 3A,B), although late presentations can occur. Some patients develop a non-progressive stable lesion that does not affect graft function, while others will require listing for retransplantation. The key finding is that the hepatic artery remains patent, indicating that the biliary injury is established at the time of implantation, although hepatic artery insufficiency due to anastomotic strictures is a recognised cause of ischaemic biliary stricturing. Hepatic artery thrombosis is a feared complication affecting 3%–5% of patients receiving a liver transplant and causes massive necrosis and collapse of the biliary tree resulting in biloma formation, which in the worst cases necessitates percutaneous drainage of the biliary tree. In all of these situations, medical management with cyclical antibiotics to treat and limit cholangitis and bile salt therapy with UDCA to promote choleuresis are the mainstays of treatment. Endoscopic biliary interventions are rarely successful for these disorders due to their diffuse nature, with efficacy rates of <30%.7 47 48 Retransplantation is often necessary in such circumstances, with endoscopic or percutaneous therapy being reserved for those few cases where external biliary drainage can be achieved and serve as a bridge towards transplantation.49 50

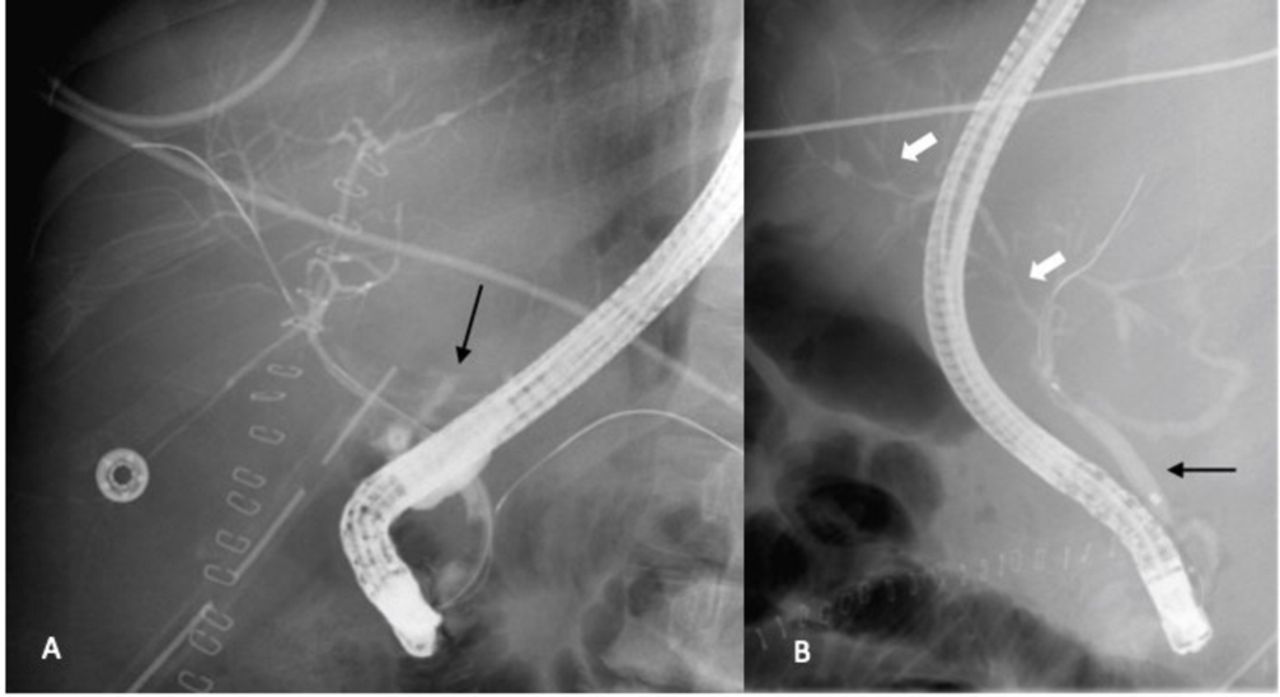
Evolution of ischaemic biliary stricturing postliver transplant. (A) A biliary leak at the site of an early anastomotic stricture (arrow) was treated with stenting. (B) One month later, after the stent was removed, the patient had developed perihilar and peripheral classic intrahepatic beading and stricturing (white arrows), typical of an ischaemic cholangiopathy. The hepatic artery remained patent. The black arrow indicates the common bile duct.
Portal biliopathy
Portal biliopathy refers to a spectrum of biliary abnormalities caused by extrahepatic portal vein occlusion (EHPVO) and is generally seen in conjunction with portal cavernoma.51–53 The development of portal biliopathy in EHPVO results from a combination of biliary ischaemia due to altered biliary vascular supply and extrinsic and intrinsic compression due to collaterals in and around the bile duct, particularly in the subhilar and perihilar regions. Over a longer period of time, thrombosis of the small bile duct venules leads to biliary inflammation and fibrosis, causing ductal strictures, cholestasis and the development of stones/sludge. Biochemical and biliary abnormalities can be seen in 80%–100% of patients with EHPVO, but only a small proportion of these patients (5%–30%) develop symptoms due to biliary obstruction.53 54
Clinical features
Symptomatic patients develop progressive cholestasis and jaundice, cholangitis, cholecystitis and abdominal pain due to cholelithiasis. Duration since the onset of EHPVO is a strong predictive factor, with a study from Birmingham, UK noting that clinical symptoms of portal biliopathy became manifest around 7 years after the onset of EHPVO.55
Investigations
Cross-sectional imaging with MRCP/CT facilitates a diagnosis and evaluation of EHPVO, in addition to delineating biliary abnormalities. EUS assists in a detailed assessment of biliary strictures and exclusion of other pathology such as malignancy or chronic pancreatitis. ERCP is useful in obtaining a detailed overview of the biliary system and concurrent abnormalities but is generally used to provide biliary decompression.
Management
Asymptomatic patients do not require any specific biliary management, except a general management of their portal hypertension with non-selective beta blockers (efficacy not proven in EHPVO) and/or variceal band ligation if appropriate. The role of anticoagulation in EHPVO is controversial and is only considered if there is an underlying prothrombotic state.56
Endoscopic therapy for portal biliopathy involves ERCP for biliary decompression (strictures) and for extraction of stones/sludge from the bile duct. Biliary dilatation and/or sphincterotomy in the setting of portal biliopathy can be risky due to the presence of choledochal varices. Plastic or fully covered self-expanding metal stents are used to treat strictures. Percutaneous biliary drainage may be necessary in some patients.
The ideal treatment for portal biliopathy is portal venous decompression with a portosystemic shunt (flow chart in figure 4). Transjugular intrahepatic portosystemic shunt may be challenging in the presence of EHPVO and portal cavernoma, with success often limited due to significant attenuation of the portovenous inflow and lack of a suitable target vein. Liver transplantation is rarely possible due to the loss of classical portal vein anatomy, and in this setting early referral for multivisceral transplantation should be made in selected cases.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
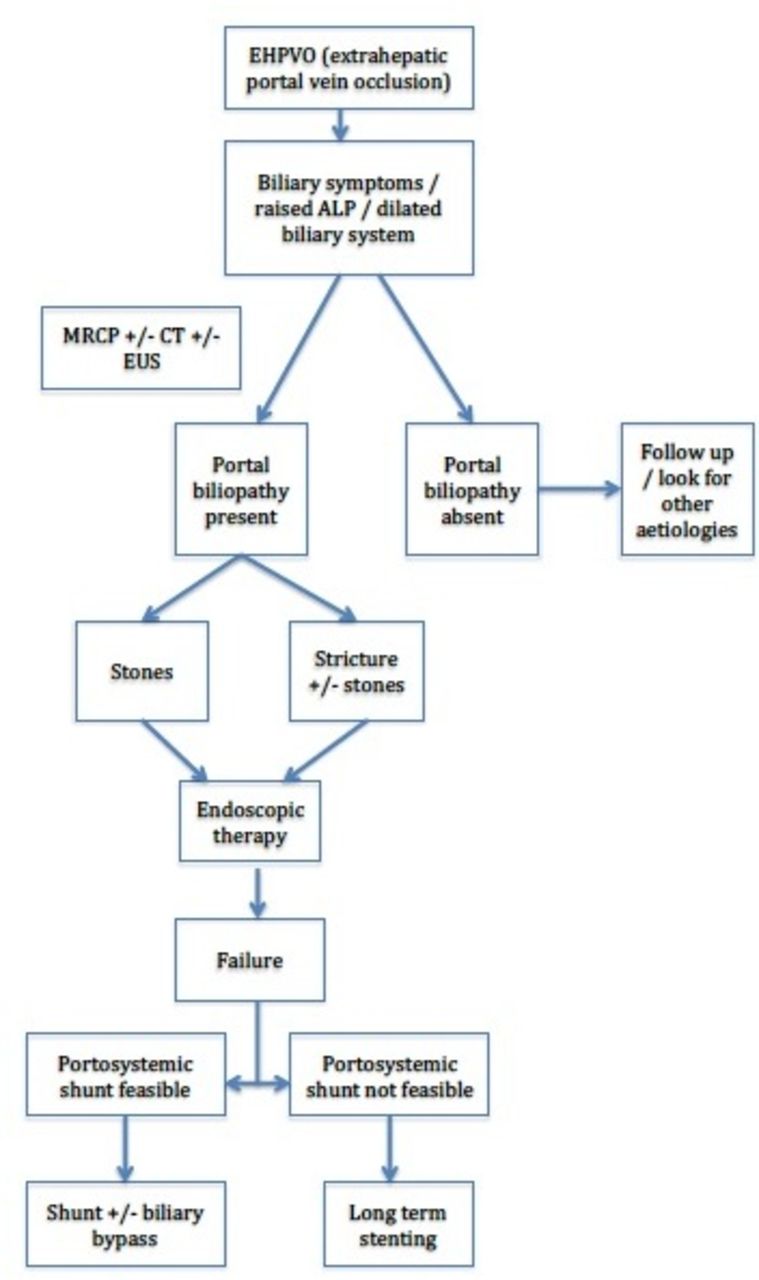
Management algorithm for extrahepatic portal vein occlusion (EHPVO). ALP, alkaline phosphatase; EUS, endoscopic ultrasound; MRCP, magnetic resonance cholangiopancreatography.
Recurring pyogenic cholangitis
Recurring pyogenic cholangitis or oriental cholangitis is a condition in which intrabiliary pigment stone formation results in recurrent bouts of cholangitis and concurrent biliary stricturing in people who live, or have lived, in South-East Asia.57 The pathogenesis of intrabiliary pigment stones is uncertain and stones are thought to occur de novo, although biliary parasitic infections due to liver trematodes (flukes) and roundworm are thought to play an important role in initiating and propagating recurrent cycles of biliary infection.58 Despite the epidemiological association of parasites, such infections are only seen in 20%–45% of patients,59 and recurrent pyogenic cholangitis is common in parts of South-East Asia where these parasites are not endemic.60
Following transient portal bacteraemia, secondary stone formation and biliary stricturing may occur. These stones tend to be composed of calcium bilirubinate in contrast to more common cholesterol stones in Western populations, and patients experience recurrent bouts of acute cholangitis and stone-associated pancreatitis, which may cause abscesses at extrahepatic sites. Treatment involves acute management of cholangitis with broad-spectrum antibiotics and ductal drainage via ERCP or percutaneous transhepatic cholangiography (PTC). Definitive management of stones is generally achieved using ERCP. Although cholangioscopy and lithotripsy may assist in clearing extrahepatic stones,61 resection of hepatobiliary segments containing significant numbers of intrahepatic stones may be necessary. Recurrent sepsis and secondary biliary cirrhosis can cause portal hypertension and increase the risk of cholangiocarcinoma, which occurs in up to 5% of patients each year.62
Caroli disease
Caroli disease is a rare disorder caused by ciliary dysfunction63 which leads to congenital ductal plate malformations resulting in saccular and non-obstructing segmental dilatations of the intrahepatic bile ducts throughout the liver.64 The typical appearance is one of intrahepatic cystic dilatations lying in continuity with the biliary tree and an otherwise normal common bile duct. The disorder is often found in association with autosomal recessive polycystic kidney disease caused by mutations in PKHD1, a gene which encodes a protein called ‘fibrocystin’ expressed in the kidney but also found in cholangiocytes.65
Two forms of Caroli disease are described—a simple form (termed Caroli disease) which is strongly associated with medullary sponge kidney, and a second more complex syndrome associated with congenital hepatic fibrosis which causes periportal fibrosis, portal hypertension and varices.66 67 Unlike choledochal cystic disease and massive hepatic cysts where there is a female preponderance, Caroli disease seems to affect men and women equally but is more commonly seen in people of Asian descent.68 69 The first presentation is usually in teenage or young adult life with symptoms of cholangitis and right upper quadrant pain and diagnosis usually made by MRCP examination of the biliary tree.
Chronically impaired biliary drainage and the presence of biliary cisterns can lead to the formation and accumulation of intrahepatic stones and sludge in up to a third of patients, which causes chronic cholangitis and jaundice. The risk of cholangiocarcinoma is significantly increased by chronic cholangitis and is thought to occur in 7% of affected individuals.70
Management is concerned with minimising the risk of cholangitis using antibiotics (sometimes rotating courses), and in stone formers long-term ursodeoxycholic acid to reduce stone formation. Endoscopic clearance of the distal bile duct can help in patients with choledocholithiasis but is of limited utility in clearing large intrahepatic stones, which may need targeted extracorporeal lithotripsy combined with PTC drainage and irrigation, while the rarer forms of limited segmental disease can be considered for hepatic lobectomy. The requirement for PTC-based therapies usually heralds the onset of more complicated disease where it can prove difficult to control cholangitis and definitive interventions such as liver transplantation should be considered.69 Liver transplantation or liver and kidney transplantation in the setting of polycystic kidney disease may be associated with a higher risk of mortality due to the risk of infection due to cyst infection and cholangitis, particularly in patients with severe Caroli disease.71
Conclusion
Large-duct cholangiopathies refer to a group of discrete conditions with varying aetiologies but common clinical presentations (mainly cholangitis). The management of such patients is challenging and will benefit from a specialist multidisciplinary approach, while the timing of biliary interventions in obstructive disease should be determined by the nature of the underlying lesion and the likelihood of biliary drainage being restored.
In a post-transplant setting, clinicians should be aware that biliary complications are common and that the underlying cause may reflect recipient as well as donor factors. Ischaemic lesions need careful work-up and careful consideration of therapeutic intervention, while early anastomotic strictures and leaks respond well to prompt endoscopic intervention.
In the future, pharmaceutical manipulation of bile salt production and the enterohepatic recirculation promises to improve management of parenchymal biliary disease.
Acknowledgments
The authors would like to acknowledge Dr P Trivedi, University of Birmingham, for sharing his slides, and Dr Richard Sturgess, Aintree University Hospitals NHS Foundation Trust, for sharing a cholangiographic image.
References
Footnotes
Contributors Both SM and AH have been involved in planning, research and preparing the manuscript.
Funding Neither SM nor AH has received any funding for this review.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Not required.