Article Text

Statistics from Altmetric.com
Compared with our ancestors, Western societies today lead a lifestyle that is much more sedentary, probably as a result of cultural changes stemming from modern socio-economic morays. Taking into account differences in body size, our energy expenditure per kilogram of body weight has been estimated to be <40% of that of our prehistoric ancestors.1 Current estimates suggest that 7 out of 10 adults are inactive or lack adequate conditioning,2 and this lack of adequate exercise, combined with dietary indiscretion, has contributed to the worldwide epidemic of obesity and non-alcoholic fatty liver disease (NAFLD). Within the USA, data suggest that 64.5% of the adult population is now overweight or obese, with a worldwide prevalence of 40–60%.3–5 Obesity, combined with host factors such as diet, sedentary lifestyle and genetic predisposition, has been directly associated with increases in the prevalence of insulin resistance, diabetes mellitus and the metabolic syndrome. The hepatic manifestation of the metabolic syndrome, NAFLD, has also increased in prevalence, now considered to be around 20–30% in Western countries. Among morbidly obese patients undergoing bariatric surgery, approximately 90% have NAFLD and 36–37% have the more aggressive form of fatty liver, non-alcoholic steatohepatitis (NASH).6 7 In patients with NAFLD, advancing age, increasing weight, the number of features of the metabolic syndrome and the degree of insulin resistance have all been independently associated with NASH severity.8 Evidence-based treatment options for NAFLD are currently lacking. Recent data suggest that the thiazolidinedione class of insulin sensitisers may be efficacious, but widespread utilisation of these agents awaits further investigation.9 Evidence also supports a role for weight loss, achieved through diet and exercise.
The purpose of this review is to describe the currently accepted pathophysiology of NAFLD, to highlight the known dietary and exercise habits of patients with NAFLD, and to detail the benefits of lifestyle modification with diet and/or exercise on parameters of metabolic syndrome, including insulin resistance and NAFLD. The effects on liver histopathology, where available, will also be detailed.
PATHOPHYSIOLOGY OF NAFLD
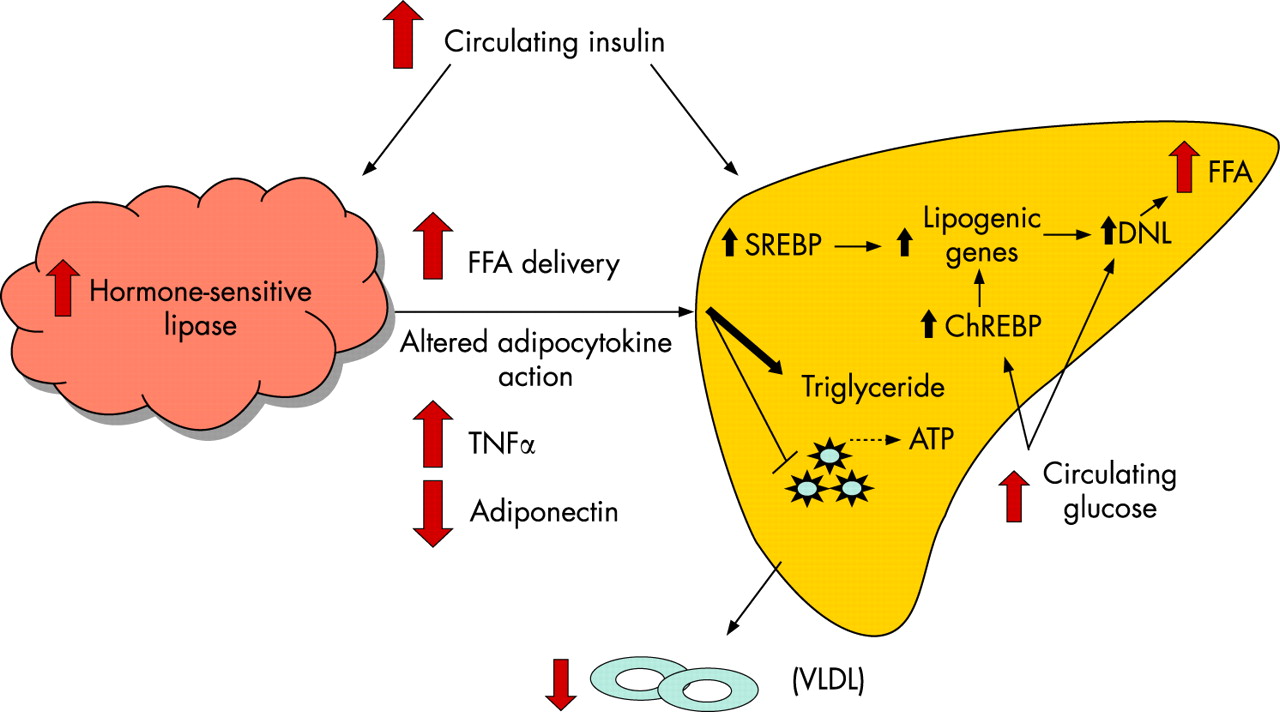
The majority of NAFLD patients are overweight or obese and have underlying insulin, and probably leptin, resistance that results in dysregulated energy metabolism. The regulation of glucose and lipid metabolism involves a complex interplay between adipose tissue, skeletal muscle and the liver. While our knowledge of the pathogenesis of hepatic steatosis has undoubtedly increased over the last decade, many uncertainties remain, and it remains the subject of intense investigation. Hepatic steatosis derives from several possible sources including: (1) increased free fatty acid (FFA) delivery to the liver as a result of dietary fat intake and increased lipolysis within insulin-resistant adipose tissue; (2) increased hepatic de novo lipogenesis (DNL); (3) decreased FFA oxidation; and (4) decreased triacylglycerol (TAG) export from the liver in the form of very low-density lipoprotein (VLDL). The largest contributor to hepatic steatosis in patients with NAFLD is increased FFA influx to the liver (60%), followed by DNL (∼26%).10
Increased hepatic lipid supply
In insulin-resistant states, principally obesity and type 2 diabetes, adipose tissue hormone-sensitive lipase activity is not fully suppressed by insulin, resulting in enhanced lipolysis and non-esterified fatty acid (NEFA) flux into the systemic circulation (fig 1). The precise mechanism of adipocyte insulin resistance in obesity remains a subject of controversy, but an emerging body of data suggest that an altered adipocytokine milieu in visceral fat resulting from macrophage infiltration is important.11 This milieu is characterised by increased expression of pro-inflammatory cytokines, such as tumour necrosis factor α (TNFα) and interleukin 6 (IL6), that can directly inhibit insulin signalling, and decreased expression of adiponectin, an anti-steatotic, and insulin-sensitising adipocyte-derived cytokine (adipocytokine) in both skeletal muscle and liver.12
Hepatic lipid synthesis and oxidation
Energy metabolism within the liver is tightly regulated. Two transcription factors, sterol regulatory element-binding protein (SREBP-1) and carbohydrate response element-binding protein (ChREBP), are intimately involved in hepatic glucose and lipid metabolism, and their activity is increased in animal models of NAFLD.13 14 The former is induced by insulin and high-fat diets, and regulates glycolytic and lipogenic gene expression resulting in increased DNL and a concomitant decrease in FFA oxidation resulting from malonyl-CoA-induced inhibition of carnitine palmitoyl transferase (CPT)-1 reducing mitochondrial FFA uptake.15 ChREBP exerts similar effects on glycolytic and lipogenic gene expression and also increases the expression of genes involved in triglyceride synthesis. In contrast to SREBP-1, ChREBP is up-regulated by glucose, which increases its nuclear translocation and its DNA binding/transcriptional activity.16 Inhibition of ChREBP in ob/ob leptin-deficient mice reduces hepatic steatosis by decreasing lipogenesis and enhancing FFA β-oxidation, with a concomitant decrease in circulating plasma triglycerides and FFA resulting in the restoration of hepatic, skeletal muscle and adipose tissue insulin sensitivity.14 This study provides further evidence linking fat accumulation to insulin resistance in both hepatic and non-hepatic tissues, principally skeletal muscle.17 18
Insulin resistance
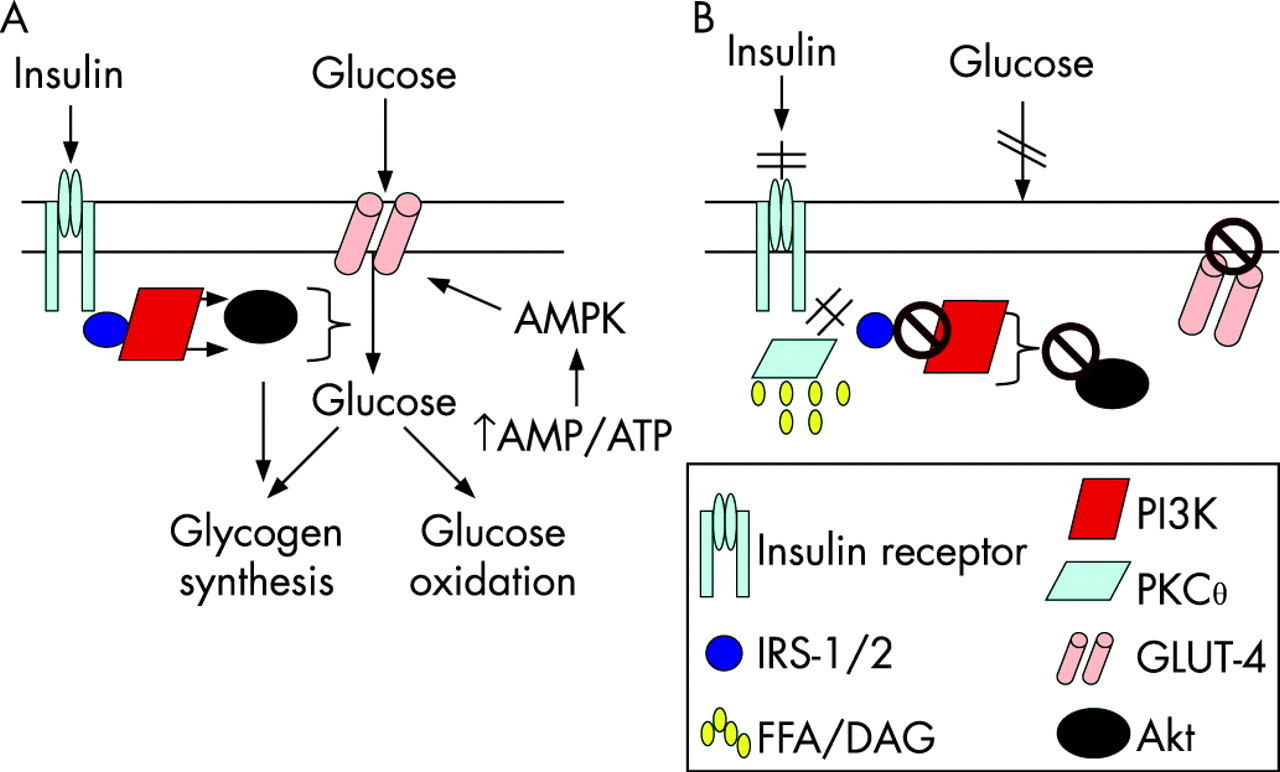
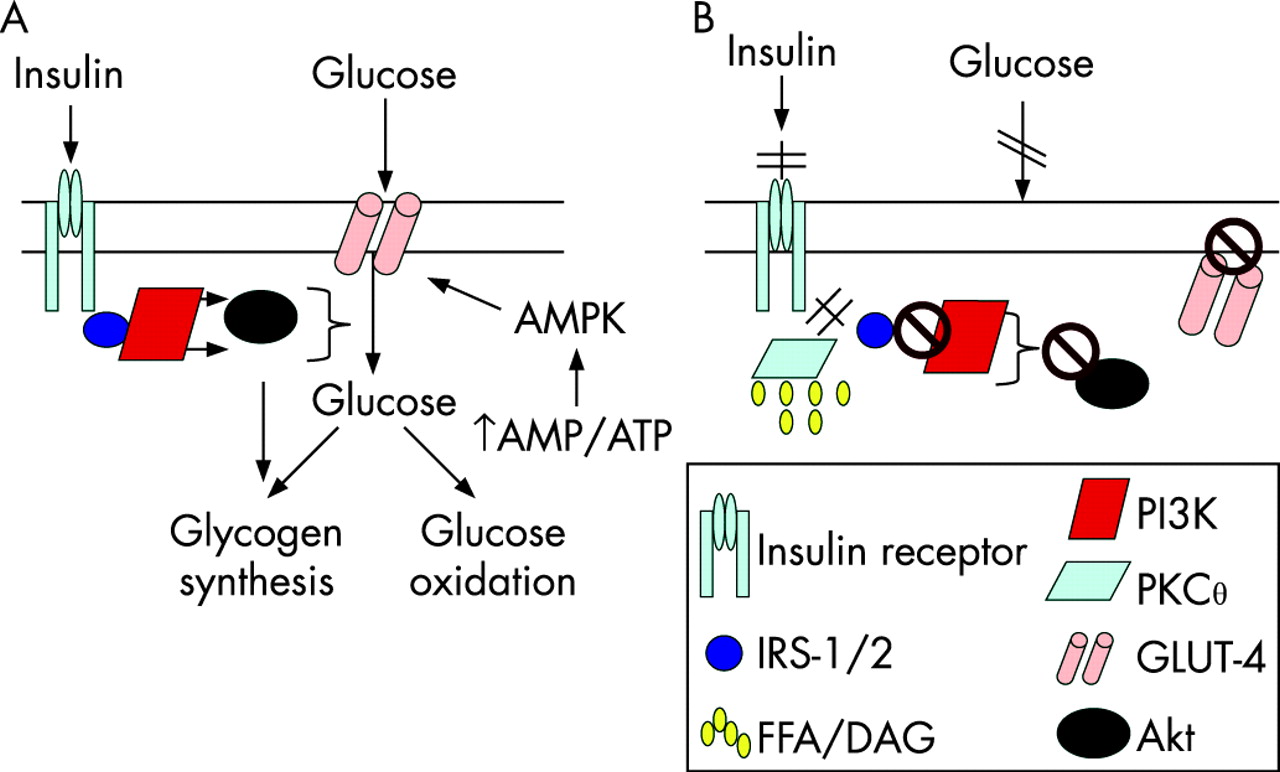
Skeletal muscle is the primary site for glucose disposal via insulin-dependent pathways, accounting for about 75% of whole-body insulin-stimulated glucose uptake.19 Insulin binding to the insulin receptor on the myocyte plasma membrane results in autophosphorylation of the receptor, allowing insulin receptor substrate (IRS)-1 adaptor protein to bind and undergo tyrosine phosphorylation (fig 2A). IRS-1-associated phosphatidylinositol 3-kinase (PI3K) activity is increased, which results in downstream activation of Akt/PKB, leading to enhanced glucose uptake into the cell, via increased GLUT-4 translocation from the cytosol to the cell membrane, and up-regulation of glycogen synthesis and glucose oxidation.18 In obese individuals, there is reduced insulin-stimulated glucose disposal in skeletal muscle, most probably due to increased intramyocellular lipid content (fig 2B).18 The increased concentration of FFA (or more probably their esterification product, diacylglycerol (DAG), activates the serine/threonine kinase PKCθ which phosphorylates IRS-1 on critical serine sites, thereby inhibiting its tyrosine phosphorylation and subsequently the activation of PI3K and the resulting glucose uptake, glycogen synthesis and glycolysis. Other cytokine-regulated serine/threonine kinases including IκB kinase-β (IKKβ) and JNK-1 may also be involved since the inhibition of IKKβ via exercise20 or salicylates results in improved insulin sensitivity.21 As discussed above, the hyperglycaemia resulting from skeletal muscle insulin resistance in obesity, will, via activation of ChREBP, lead to increased hepatic fat synthesis, reduced fat oxidation and steatosis. Moreover, the accumulation of FFA/DAG in the liver results in hepatic insulin resistance, via PKC∊-induced IRS-2 serine phosphorylation, which, reduces insulin’s inhibitory effect on gluconeogenesis, contributing further to hyperglycaemia.18 22
{kind=link}
{kind=link}
Leptin resistance
Lack of response to the adipocytokine leptin (leptin resistance) rather than insulin resistance may be an important factor in the pathogenesis of hepatic steatosis. The evidence for leptin resistance in NAFLD, though indirect, is compelling. Leptin exerts a number of anti-steatotic effects on the liver, including inhibition of lipid synthesis via reduced expression of stearoyl CoA desaturase (SCD)-123 and enhanced FFA oxidation, via up-regulation of peroxisome proliferator-activated receptor α (PPARα),24 and yet patients with NAFLD have increased serum leptin concentrations.25 Recent studies in genetically leptin-resistant (ZDF) rats have dissociated the direct anti-steatotic effects of leptin from any indirect effects via improved hepatic insulin sensitivity.26 The mechanism of leptin resistance in obesity is unclear; however, it may be enhanced by the ingestion of fructose which leads to an inhibition of STAT-3, abrogating leptin-mediated PPARα activation resulting in decreased fatty acid oxidation, increased SREBP-1 expression and increased hepatic triglyceride content.27 Further inhibition of STAT-3 may occur as a result of up-regulation of suppressor of cytokine signalling-3 (SOCS-3) by adipose tissue or hepatocyte-derived inflammatory cytokines.15 28
AMP-activated protein kinase
Of particular relevance to the therapy of NAFLD, AMP-activated protein kinase (AMPK) has recently emerged as a key orchestrator of both hormonal and non-hormonal regulators of energy metabolism in liver, skeletal muscle and adipose tissue. Activated by an increase in the AMP/ATP ratio, AMPK activity is enhanced by physiological processes that induce metabolic stress and either decrease ATP production (ischaemia, hypoxia) or increase its consumption (exercise).29 Once activated, AMPK acts to increase ATP-generating cellular events while turning off energy-consuming processes to restore energy balance. In the liver, the activation of AMPK by starvation, exercise,30 adiponectin,31 leptin (via inhibition of SCD-1), the biguanide metformin32 or the thiazolidinedione class of insulin sensitisers33 suppresses expression of SREBP-1,34 ChREBP,35 and acetyl-CoA carboxylase (ACC),36 resulting in decreased DNL and increased FFA oxidation. Hepatic gluconeogenesis is also inhibited. Within skeletal muscle, exercise, leptin or drug-related AMPK activation enhances glucose uptake via direct non-insulin-dependent GLUT-4 translocation, and increases pyruvate oxidation.32 In adipose tissue, hormone-sensitive lipase activation is suppressed, resulting in decreased lipolysis.32
Decreased hepatic lipid export
Hepatic fat, in the form of TAG-rich VLDL, can be transported out of the liver. This occurs, in part, via microsomal triglyceride transfer protein (MTP)-mediated TAG association with apolipoprotein B (ApoB). Inhibition of MTP in mice results in inefficient lipidation of ApoB, leading to a lack of TAG transfer out of the liver.37 Hyperinsulinaemia can alter the synthesis of ApoB, leading to decreased VLDL production,38 and hyperinsulinaemic NASH patients have been reported to have impaired synthesis of ApoB, almost certainly contributing to hepatic fat accumulation.39
Inflammation and fibrosis
The precise mechanisms leading to inflammation and subsequent fibrosis in NAFLD are far less clear than those involved in steatosis; however, most available evidence suggests that many of the factors contributing to fat accumulation also play a role in progressive disease.40 This implies that any lifestyle modifications aimed at reducing steatosis should also improve inflammation and fibrosis. As an example of these common “factors”, an increased hepatic supply of FFAs in the face of hepatic insulin resistance will favour increased mitochondrial FFA β-oxidation and the subsequent generation of oxidative stress. Oxidative stress, along with other FFA-induced mechanisms including lysosomal cathepsin release and endoplasmic reticulum stress, leads to the transcription and release of pro-inflammatory cytokines from hepatocytes, which will add to the pro-inflammatory milieu resulting from adipose tissue inflammation and further inhibit the release and actions of the anti-steatotic, anti-inflammatory and anti-fibrotic adipocytokine adiponectin. Potential non-inflammatory mechanisms of fibrosis in NAFLD include the direct pro-fibrotic effects of leptin, insulin, norepineprine and angiotensin, all released in increased amounts either from adipose tissue in obesity or, in the case of insulin, by the pancreas in response to obesity-related insulin resistance.
NUTRITION AND EXERCISE IN PATIENTS WITH NAFLD
The dietary and exercise habits of patients with NAFLD vary based on ethnicity, geography and culture. However, perhaps not surprisingly, available data consistently show that energy intake is significantly higher in patients with NAFLD than in individuals with no evidence of fatty liver.41 In the USA, high-fructose corn syrup ingestion has increased dramatically over the past 20 years42 and may contribute to the underlying dysregulation in energy metabolism seen in NAFLD patients. High-fructose diets in humans are associated with increased energy intake, weight gain, increased fat mass and increased blood pressure after only 10 weeks.43 They have also been associated with increased hepatic DNL, hypertriglyceridaemia and the development of hepatic insulin resistance.44 Animal data have shown that these processes are probably the result of fructose-mediated leptin resistance resulting in decreased hepatic fatty acid oxidation via reduced leptin-mediated PPARα activation.27 Musso et al studied the dietary intake of 25 NASH patients over a 7-day period, compared with 25 age-, body mass index (BMI)- and gender-matched controls.45 Patients with NASH were found to have diets higher in saturated fat and cholesterol and lower in polyunsaturated fat, fibre and anti-oxidant vitamins C and E. Cortez-Pinto et al also showed that NASH patients recalled diets higher in total and monounsaturated fat, as well as n-6 fatty acids.46 Animal and in vitro data suggest that saturated and trans-unsaturated FFAs increase insulin resistance while polyunsaturated FFAs exert the opposite effect.47 Polyunsaturated FFAs may also be directly anti-steatotic via inhibiting the abundance and DNA binding activity of SREBP-1.48 Consistent with the activation of ChREBP by glucose, two studies have recently reported an association between dietary carbohydrate content and NAFLD severity.8 49
In addition to poor dietary habits, lack of physical exercise adversely impacts metabolic profiles significantly. Physical inactivity is associated with increased body weight, central adiposity and insulin resistance,50 and increased risk of metabolic syndrome,51 NAFLD52 and severity of NASH.8 Many of these effects may be mediated by low levels of AMPK activation.30
DIETARY MODIFICATIONS FOR NAFLD
The primary problem in the majority of patients with NAFLD is overweight or obesity and underlying insulin resistance. It therefore seems logical that dietary advice would form the cornerstone of therapy for these patients. From the above discussion, diet-induced weight loss would be expected to have a number of inter-related beneficial effects on NAFLD broadly related to (1) reduced hepatic FFA supply; (2) improved insulin sensitivity; and (3) reduced adipose tissue inflammation (table 1). Unfortunately, large, prospective, randomised clinical trials powered appropriately for histopathological end-points are lacking. Rather, several small, uncontrolled, pilot trials utilising differing caloric restriction regimens and combinations of carbohydrate, protein and lipid have been performed, reporting on a wide variety of histological and non-histological end-points (table 2).
General caloric restriction
One of the first prospective studies to assess the effect of diet-induced weight loss on liver pathology was performed in 1970 by Drenick et al.53 In this trial, three different weight loss regimens were evaluated in 41 severely obese patients. The majority of patients were treated with either prolonged fasting or a 500 kcal/day diet. Patients lost a mean of 40.9 kg during prolonged fasting for a mean of 71 days and 59.5 kg over an average of 5 months in the low calorie arm. Repeat liver biopsies showed improvement in hepatic steatosis. In 14 patients who were able to maintain the weight loss for an average of 17 months, another biopsy was performed which showed normalisation of histology in nine patients and minimal abnormalities in the remaining five. A subsequent small retrospective cohort of obese patients demonstrated that a 10% weight reduction achieved on a 600–800 kcal/day diet for a mean of 16 months resulted in improvement in liver enzymes and hepatosplenomegaly; however, no follow-up liver biopsies were performed.54
Specific macronutrient restriction
The dual role of lipid and carbohydrate as both providers of substrates for hepatic lipid synthesis and activators of SREBP-1 and ChREBP transcription factors has prompted studies examining their specific restriction in combination with calorie-restricted diets in patients with NAFLD. The effect of a low-fat (3%) reduced-calorie (daily intake 1200 kcal/day) diet was studied by Petersen et al in eight patients with NAFLD.55 An average 8% weight loss was associated with an 81% reduction in intrahepatic lipid content as measured by magnetic resonance proton spectroscopy (MRS). Whole-body insulin sensitivity also improved, and hepatic triglyceride content, fasting plasma glucose and total cholesterol were significantly reduced. Utilising deuterated glucose, the authors demonstrate that improvement in whole-body insulin sensitivity was predominantly due to improved hepatic insulin responsiveness. Westerbacka et al examined the effect of an isocaloric, low-fat (16%) diet followed by an isocaloric high-fat (56%) diet on hepatic lipid content determined by MRS, in a 2-week crossover study in 10 obese non-diabetic women.56 The low-fat diet led to a 20% decrease in hepatic lipid content which was followed by a 35% increase on the high-fat diet. Reports that low-carbohydrate, ketogenic diets (<20 g/day) are associated with a greater weight loss and a better lipid profile than low-fat diets57 have prompted a recent pilot trial of 6 months on a low-carbohydrate diet in five obese NAFLD patients.58 A mean weight reduction of 12.8 kg was associated with a significant improvement in hepatic steatosis and inflammation, and a trend towards an improvement in fibrosis. As yet, only one dietary study has specifically targeted patients with biopsy-proven NASH.59 In this open label study, 16 NASH patients were placed on a diet of 40–45% complex carbohydrate/fibre, 35–40% fat (mainly mono- or polyunsaturated fat) and 15–20% protein for 12 months, aimed at weight reduction and improved insulin sensitivity. Of the 15 patients who underwent repeat biopsies, nine had improved histology and six remained stable. The improved patients lost significantly more weight (7%) than the stable patients.
Diet plus weight-reducing pharmacotherapy
Despite these encouraging data that diet-induced weight loss may be beneficial for patients with NAFLD, the majority of patients have difficulty achieving and maintaining weight loss. Accordingly, pharmacological agents to augment dietary advice have begun to be evaluated as therapy for patients with NAFLD. Orlistat, an oral inhibitor of gastric lipases, blocks the absorption of approximately 30% of dietary triglycerides. Early data, in the form of a case series, showed that orlistat 120 mg three times daily, in association with a low-fat diet, induced weight loss of 10–19 kg over a 6–12 month period and was associated with a significant improvement in serum aminotransferases and hepatic steatosis, inflammation and fibrosis.60 This report was followed by an open label trial of orlistat 120 mg three times daily plus a low-fat diet in 10 obese patients with biopsy-proven NASH. The mean weight loss was 10.3 kg. Significant improvements in BMI, haemoglobin A1C, serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were seen. Histopathological improvement in fibrosis was found in three of five patients who were able to lose at least 10% of their body weight.61
More rigorous controlled trials with orlistat have since been published. Zelber-Sagi et al studied the effects of orlistat in 52 patients randomised to receive placebo or orlistat 120 mg three times daily for 6 months.62 Both groups were prescribed a low-energy diet consisting of 104.5 kJ/day and encouraged to walk for 40 min, 3–4 times weekly. Among the 44 patients who completed the study, BMI was significantly reduced, with no difference found between groups. The mean weight loss was 8% in the orlistat group and 6% in the placebo group. Serum ALT was reduced significantly in both groups, with a greater decrease in the orlistat group. However, utilising ultrasound assessment of hepatic steatosis, reversal of steatosis was seen only in the orlistat group. Similar findings have recently been reported from a second randomised controlled trial of orlistat in 50 overweight, biopsy-proven NASH patients treated with a 1400 kcal/day diet or a similar diet plus orlistat 120 mg three times daily for 9 months.63 Of the 37 patients who have completed the trial, a mean weight loss of 8.2 and 6% was seen for the orlistat and diet alone group, respectively. Both groups had similar improvements in steatosis, necroinflammation, ballooning and NAFLD activity score (NAS) on follow-up biopsy. When all patients from both groups losing ⩾9% of body weight (n = 14) were compared with those failing to achieve this amount of weight reduction, greater improvements in insulin resistance, AST, adiponectin, steatosis, inflammation and NAS were seen in the >9% group. However, no improvement in fibrosis was shown. While confirming the benefits of diet-induced weight loss on NAFLD, these two studies show that orlistat does not significantly enhance weight loss induced by a hypocaloric diet, a finding corroborated by a recent Finnish study in obese women.64
Intensity of weight loss
Data from some early studies suggested that too rapid a weight loss may worsen the histopathology of patients with NAFLD. Anderson et al evaluated the effects of a 388 kcal/day diet on 41 severely obese patients who lost a median of 34 kg over about 9 months.65 While steatosis significantly improved, 24% developed slight portal inflammation or slight fibrosis. No patients losing <1.6 kg/week had a worsening of fibrosis. This, along with early bariatric surgery data demonstrating an increase in inflammation following a mean of 32 kg weight loss,66 led some investigators to advocate slow and mild to moderate weight reduction for patients with NAFLD. More recent longer term follow-up data from bariatric surgery studies have, however, largely allayed these concerns,67–69 suggesting that the earlier reports may have been attributable either to poor nutritional support in these patients or to a transient histopathological phenomenon occurring en route to resolution of NAFLD.
Taken together, these data demonstrate that diet-induced weight loss approaching 10% of body weight is efficacious in improving the metabolic parameters associated with NAFLD, including lipid profiles and insulin resistance, as well as serum aminotransferases, and, more importantly, the histopathological characteristics of NASH including steatosis and necroinflammation. Further study is necessary to determine if weight loss over a longer period of time equates to improvement in fibrosis in patients with NASH. Additionally, no consensus has been reached as yet on the most appropriate nutrient balance in diets for NAFLD patients. A low processed carbohydrate diet may achieve a more rapid weight loss, but this effect is probably mitigated in the long term.70 To date no adjunctive weight-reducing pharmacological therapies have, as yet, been shown to be of any benefit.
EFFECT OF EXERCISE ALONE ON OBESITY, VISCERAL FAT AND INSULIN RESISTANCE
Exercise physiology and the salutatory effects on weight loss, fat reduction and insulin sensitivity have been described in great detail. These beneficial effects are now considered to reflect, at least in part, the effect of exercise on the activation of AMPK. In obese non-diabetics, exercise has been shown to reduce the risk of developing diabetes mellitus by up to 46%.71 Physical training, consisting of 20 min cycling or running, 20 min swimming at submaximal heart rate, followed by 20 min of warm up/cool down three times per week for 4 weeks, resulted in a significant reduction in body weight and percentage body fat, and this was associated with improved whole-body glucose uptake, decreased fasting insulin concentrations and increased circulating adiponectin and mRNA expression in muscle. Among patients with type 2 diabetes mellitus, increasing exercise led to a reduction in fasting plasma glucose.72
The intensity of exercise needed to show improvement in metabolic profiles has been studied by several investigators. O’Donovan and colleagues evaluated the effects of 24 weeks of moderate intensity exercise, defined as cycling three times weekly at 60% Vo2max to burn 400 kcal, versus high-intensity exercise, defined as cycling three times weekly at 80% Vo2max to burn 400 kcal, versus no exercise, on insulin sensitivity, triglycerides and glucose concentration.73 Training at 60% Vo2max was as effective as training at 80% Vo2max when 400 kcal were expended per session, suggesting that moderate exercise, expending 400 kcal per session, three times per week is sufficient to improve insulin sensitivity. The overall energy expenditure achieved per work-out session appears to be more important than the intensity of the exercise. This is supported by two recent studies performed in obese patients.74 75 Daily exercise for 12 weeks, performed at not greater than 70% Vo2max (∼80% maximum heart rate) on a treadmill to achieve 700 kcal energy expenditure (∼60 min) resulted in an 8% body weight loss and was associated with significant reductions in abdominal obesity, visceral fat, waist circumference and insulin resistance.74 Furthermore, this study showed that exercise without weight loss also reduced both abdominal and visceral fat. O’Leary et al also demonstrated that daily aerobic exercise for 50–60 min, starting at 60–65% maximum heart rate and increasing to 80–85% maximum heart rate (∼70% Vo2max) over 4 weeks improved visceral fat content and this correlated with improved glucose metabolism and loss of insulin resistance.75 These encouraging results were seen with only a 3% weight loss over this time period.
The effects of aerobic versus restrictive exercise have also been debated. A recent Turkish study examined the effects of aerobic exercise, defined as walking briskly for 15 min and exercising on a stationary bicycle for 12–15 min three times per week the first month, exercising 20–30 min four times per week the second month, and 30–45 min five times per week the third month, in a group of 20 obese women compared with a restrictive exercise utilising a stationary exercise unit in a similar group of women over a 3-month period.76 While improvement in BMI, fasting glucose and post-prandial glucose were seen in both groups, reduced fat mass (as measured by bioelectric impedence), decreased low-density lipoprotein and insulin resistance were seen only in the aerobic exercise group. In another study in 39 older obese men, aerobic or restrictive exercise training 3 days per week for 6 months resulted in similar improvement in whole-body glucose disposal.77
EFFECT OF EXERCISE PLUS DIET ON OBESITY, VISCERAL FAT AND INSULIN RESISTANCE
The combination of diet and exercise appears to have a synergistic beneficial effect on metabolic profiles. Weight loss of ⩾5% body weight achieved with diet and exercise has been associated with a 59% reduction in the odds of developing metabolic syndrome and increasing to 83% in patients losing ⩾10% of body weight.78 Tamura et al randomised 14 type 2 diabetic patients to either a 27.9 kcal/kg/day diet or a similar diet plus exercise (walking 2–3 times/day for 30 min, 5–6 days per week) for 2 weeks.79 Diet plus exercise resulted in improved muscle insulin sensitivity as measured by a decrease in intramyocellular lipid content and improved glucose infusion rates, an effect not seen with diet alone. However, both groups decreased intrahepatic lipid concentrations by 27%. When exercise, either aerobic or restrictive, was added to a low-calorie diet in obese men, insulin levels obtained via an oral glucose tolerance test (OGTT) were approximately 2-fold better than with diet alone.80 Data from the Diabetes Prevention Program in the USA extended these findings, showing that a low-calorie, low-fat diet with moderate physical activity such as brisk walking for 150 min per week reduced the incidence of diabetes by 58%.81 Importantly, this reduced incidence of diabetes was significantly better than that achieved with the insulin-sensitising medication metformin.
EFFECT OF EXERCISE PLUS DIET ON NAFLD
A logical approach to improving the histopathological changes seen in NAFLD and NASH, based on data detailed in this review, would be lifestyle modifications that combine an exercise programme with a tailored dietary regimen (table 3). Animal data demonstrate that exercise training started simultaneously with a high-fat diet prevents the development of hepatic steatosis.82 A recent study by Baba et al evaluated 65 patients with NAFLD over a minimum of 3 months who were placed on an aerobic exercise regimen and a specific diet.83 The exercise regimen consisted of brisk walking, jogging or rhythmic aerobic exercises for a minimum of 45 min, 5 days per week, to achieve a target heart rate of 60–70% of their maximal heart rate. The dietary regimen was predicated on a total of 25 kcal/kg per day containing 60% carbohydrate, 20% fat, 20% protein and 200 mg of cholesterol. A total of 44 patients complied with the exercise programme and were included in the analysis. There was a significant improvement in BMI, waist circumference, waist to hip ratio and serum aminotransferases in the patients adherent to the diet and exercise regimen. Interestingly, those patients who exercised and did not lose weight still had a significant improvement in their aminotransferase levels. Ultimately, a 4–4.5% decrease in weight resulted in a 50% reduction in serum ALT. Others have demonstrated that a combination of diet and exercise, leading to an approximate 5% weight loss over 3 months, is associated with improved serum ALT and AST.84 A similar amount of weight loss was associated with a 3.6-fold increased probability of normalising serum ALT in a large Japanese longitudinal cohort study. In this study, patients who were able to maintain their 5% weight loss had a 4.6-fold increased likelihood of maintaining a normal ALT.85 Reductions in subcutaneous and intra-abdominal fat have also been shown with 4% body weight loss when diet and exercise regimens are combined.86
Two studies have assessed the effects of diet and exercise on histopathology in patients with NAFLD.87 88 Ueno et al demonstrated that a decrease in BMI of 3 points over a 3-month period improved hepatic steatosis.87 Hickman et al subsequently showed that steatosis and fibrosis improved in a small group of NASH patients who lost approximately 4% of their body weight over a 3-month period.88
SUMMARY
Excessive foodstuffs, particularly in the form of complex carbohydrates, increased fructose consumption and saturated fats, combined with a sedentary lifestyle have contributed to the epidemic of obesity, dysregulated metabolism and NAFLD. Our knowledge of the pathological consequences of poor dietary choices and lack of adequate exercise on adipose tissue, skeletal muscle and the liver is improving, and this will help in establishing more specific guidelines for the proper nutrient intake and exercise regimens that will improve underlying metabolic pathways and ultimately decrease the incidence and severity of NAFLD. For now it seems that diets aimed at reducing total daily energy intake, based on either a low processed carbohydrate, or low-fat macronutrient platform, aimed at achieving about a 10% weight reduction, will improve both metabolic and histopathological abnormalities in a diverse group of NAFLD patients. Moderate exercise, preferably a combination of aerobic and restrictive, performed 3–4 times per week, expending about 400 calories each time seems adequate to augment improvement in the metabolic profiles of patients with NAFLD.
More rigorous, controlled studies, of longer duration and defined histopathological end-points comparing diet alone, exercise alone and diet plus exercise are needed before better, evidence-based lifestyle modification guidelines can be established, since several questions remain unanswered. Does lifestyle modification work equally well in men versus women? Do younger patients respond better than older patients to lifestyle modification? Is there a diversity of response among various ethnic groups or in patients with fatty liver alone compared with patients with more progressive disease? Do diabetic patients with NAFLD have a better histopathological response to lifestyle modification than insulin-resistant patients without diabetes and, if so, does the same amount of weight loss equate to histopathological improvement, or do these patients need to lose more or less weight, or simply need to exercise more? Finally, are there different lifestyle modification approaches, i.e. diet alone versus diet and aerobic exercise, that work better for different patient populations?
REFERENCES
Footnotes
The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or reflecting the view of the Department of the Army or the Department of Defense.
Competing interests: CPD has acted as a consultant for several pharmaceutical companies interested in developing drugs for the treatment of fatty liver disease: Sanofi-Aventis, Astellas and Pfizer. SAH has acted as a consultant for several pharmaceutical companies interested in developing drugs for the treatment of non-alcoholic fatty liver disease: Sanofi-Aventis and Schering-Plough.