Article Text

Abstract
Inflammatory bowel disease (IBD) is an idiopathic long-term relapsing and remitting disorder including ulcerative colitis and Crohn’s disease. The aim of therapy is to induce and maintain remission. Anti-TNF therapies dramatically improved clinical outcomes but primary failure or secondary loss is a common problem as well as potential side effects potentially limiting efficacy and long-term use. The advent of new targeted agents with the potential for greater safety is welcomed in IBD and offers the potential for different agents as the disease becomes refractory or even combination therapies to maximise effectiveness without compromising safety in the future. More data are required to understand the best positioning in pathways and longer-term safety effects.
- IBD
- CROHN'S DISEASE
- ULCERATIVE COLITIS
Statistics from Altmetric.com
Introduction
In the past decade, inflammatory bowel disease (IBD) has emerged as a public health challenge worldwide with westernised nations having a growing incidence and prevalence of both ulcerative colitis (UC) and Crohn’s disease (CD). In North America and Europe, over 1.5 million and 2 million people suffer from the disease, respectively.1 Both are chronic idiopathic inflammatory disorders of the entire gastrointestinal tract. They usually have a relapsing-remitting course that require lifelong treatment to maintain remission.2
For many years, IBD has been managed with corticosteroids, aminosalicylates and immunosuppressants (ie, thiopurines). The advent of biological therapies (anti-TNF-α agents), which is the current practice, has significantly improved the outcome of patients with IBD in terms of prolonged clinical remission, corticosteroid sparing, achievement of mucosal healing and prevention of disease-related complications. However, primary failure or loss of response to biologics occur in about 50% of patients treated with these drugs. Additionally, recent advances in the management of IBD have led to a paradigm shift in the treatment goals, from targeting symptom-free daily life to shooting for mucosal healing, which eventually calls for new therapeutic strategies. Also, their safety profile, especially the increased risk of infections and cancer has always been a concern for these patients, especially during the COVID-19 era, when our patients are more concerned for immunosuppression than ever.3
Cost-effectiveness had been also important, with a constant effort to decrease the cost and improve the availability of the existing agents. Biosimilars have luckily been proven to show the same efficacy with significantly lower cost.4
The need for intravenous administration in a hospital setting is rather costly for our Health Systems and can become stressful for our patients, especially those shielding during the COVID-19 period or with mobility issues or those with a busy schedule that would not prefer to commit to a scheduled frequent hospital appointment, like students or overseas.3
Easily administered orally or subcutaneous (SC) agents that can be provided from a Homecare Company had become an option and there is an increasing demand for more of these drugs to emerge. This can in turn increase compliance and improve efficacy.
Furthermore, increased understanding of the immunopathology of IBD has led to the development of targeted therapies and has unlocked a new age in IBD management and a hope for customised treatments.2 Ultimately, the need for new effective treatments for such patients has critically emerged as an urgent priority, especially during the COVID-19 period.5
IBD likely results from microbial dysbiosis, genetic and environmental factors that disturb the gut homoeostasis, leading to immune cell activation. CD has an exaggerated predominantly T helper (Th) 1 cell response with high interferon (IFN)-γ and interleukin (IL)-12 and UC mainly Th2 response dominated by the production of IL-5 and IL-13, resulting in tissue damage.6 7
Novel agents target innate and adaptive immune pathways. Adaptive immunity through differentiation of naïve CD4 +cells to effector Th1, Th2, Th17 cells causes chronic inflammation. Diverse therapeutic options are emerging, involving small molecules, apheresis therapy, improved intestinal microecology, cell therapy and exosome therapy. In addition, patient education partly upgrades the efficacy of IBD treatment.8
In this review, the latest progress in IBD treatment is summarised to understand the advantages, pitfalls and research prospects of different drugs and therapies and to provide a basis for the clinical decision and further research of IBD. Several studies with novel drugs have shown promise in IBD including innate cell microbial sensing and toll-like receptor 9 (TLR-9) modulation, Janus kinase (JAK)-STAT pathway inhibition, selective lymphocyte trafficking inhibitors, anti-integrin therapy, IL-12/23 inhibitors, sphingosine-1-phosphate receptor-1 (S1P1R) selective agonist, miR-124 expression inhibitor, IL-10 inhibitor and PDE-4 inhibitors.7–9
Methods
A comprehensive search using four databases, PubMed, Medline, Scopus and ClinicalTrials.gov was performed using the Preferred Reporting Items for Systematic Reviews and Meta-Analyses method (figure 1). The key words ‘novel’, ‘treatment’, ‘biologics’, ‘IBD’, ‘Crohn’s’ and ‘UC’ were used. Further filtration for the existing studies to include clinical trials, reviews and meta-analysis only was used. The results were limited to those performed on the last 5 years. Animal studies and studies which were discontinued or failed were excluded. Total number of reports included on this review were 44.
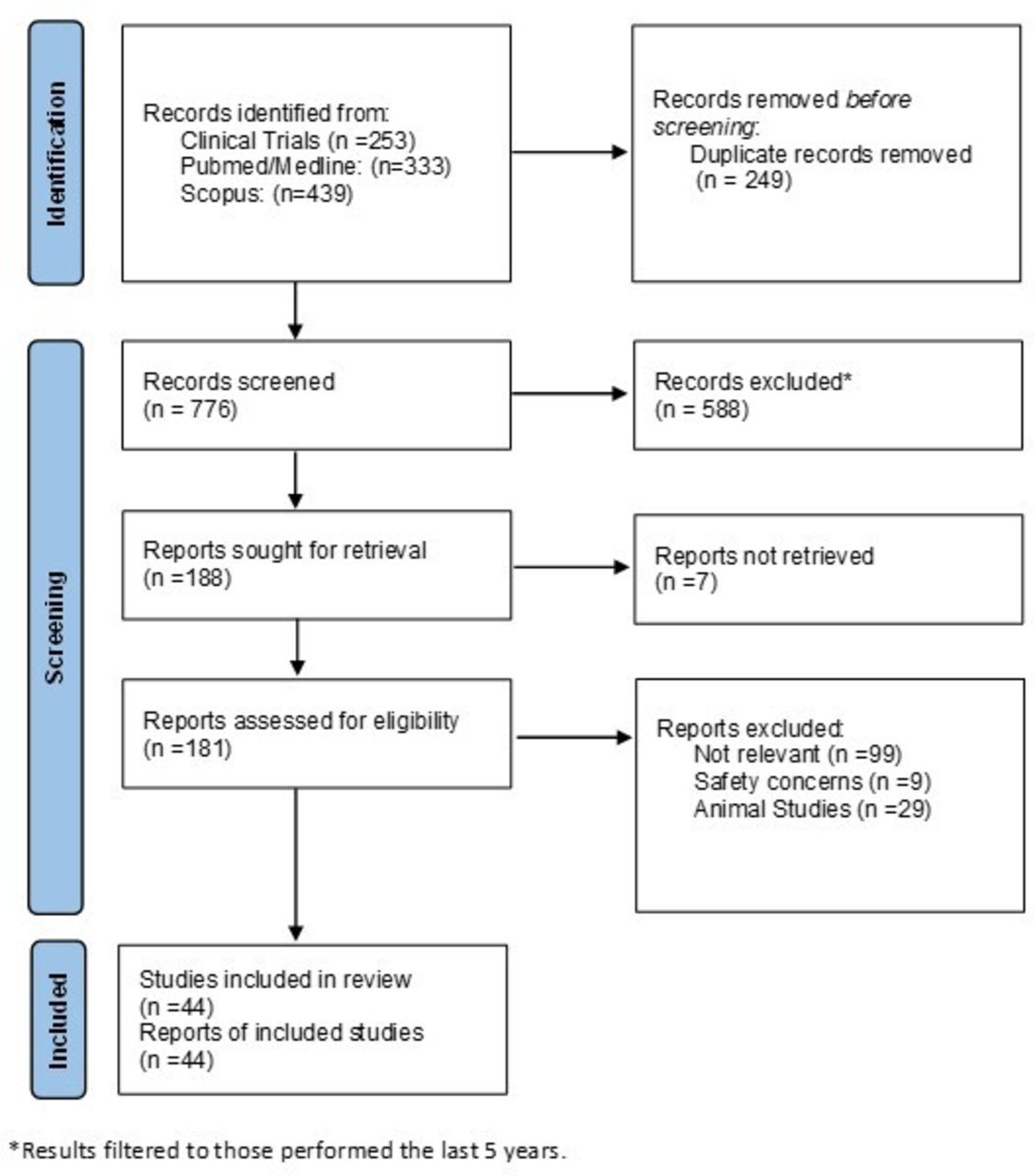
{kind=link}
PRISMA flow chart. PRISMA, Preferred Reporting Items for Systematic Reviews and Meta-Analyses.
Novel therapies
JAK-STAT inhibitors
The JAK family consists of four intracellular proteins (JAK 1,2,3 and tyrosine-kinase TYK2) associated to cytokine receptors, which mediate signal transduction on ligand binding, culminating in activation of transcription factors (STAT proteins) and gene expression via JAK-STAT pathway. JAK inhibitors block multiple signalling pathways interfering with production of cytokines and differentiation of effector T-cells.7 Tofacitinib licensed in UC belongs to this group.
Upadacitinib (UPA) is a selective JAK1 inhibitor. In the U-ACHIEVE phase IIΙ double-blind randomised controlled study (RCT) for moderate to severe UC, significantly higher proportion of patients receiving UPA 45 mg once daily) (26.1%) vs placebo/PBO (4.8%) achieved clinical remission at week 8 (p<0.001). Endoscopic remission was achieved in 13.7% vs 1.3% of placebo(p<0.001). A significant difference in clinical response favouring UPA versus PBO was seen as early as week 2 (60.1% vs 27.3%) and sustained over 8 weeks (79.0% vs 41.6%). The most common adverse effects were acne, creatine phosphokinase elevation and nasopharyngitis with UPA and worsening of UC and anaemia with PBO. Incidence of serious infection was similar between UPA and PBO. Neutropenia and lymphopenia were reported more frequently with UPA vs PBO. No adjudicated gastrointestinal perforation, major cardiovascular AEs, or thrombotic events and no active tuberculosis, malignancy, or deaths were reported.10 11
In U-ACCOMPLISH randomised controlled phase III trial, 8-week UPA 45 mg once daily has induced clinical remission in 33.5% of patients with UC, vs 4.5% of placebo, endoscopic remission in 44% vs 8.3% and histological remission in 36.7% vs 5.8% (p<0.001) induction treatment led to statistically significant improvements in clinical, endoscopic and combined endoscopic-histological endpoints. The treatment was well tolerated, and the safety profile and adverse effect prevalence was comparable with previous studies of UPA with no new safety signals identified.12
In the CD CELEST phase II trial, UPA did not significantly improve clinical remission at week 16 at any dose; however, endoscopic remission at week 12/16 was increased compared with placebo in a dose-dependent manner. Adverse events in both studies were consistent with prior reports including infections, viral reactivation and a case of pulmonary embolism and deep vein thrombosis.13 However, in preliminary results from U- EXCEED phase III RCT in patients with CD, 39% of patients achieved remission vs 21% of placebo at week 12.14
Oral JAK1 inhibitor filgotinib 200 mg daily in FITZROY CD phase 2 trial of 174 patients, was superior to placebo for induction of clinical remission (47% at 10 weeks vs 23% placebo group; p=0.0077). In phase 2b/3 SELECTION UC trial, the group receiving filgotinib 200 mg had clinical remission than placebo at week 10 (induction study A 26.1% vs 15.3%, p=0.0157). At week 58, 37.2% of patients given filgotinib 200 mg had clinical remission vs 11.2% placebo (p<0.0001). Clinical remission was not significantly different between filgotinib 100 mg and placebo at week 10 but was by week 58 (23.8% vs 13.5%, p=0.0420). Incidence of serious adverse events/adverse events was similar in both groups. Filgotinib 200 mg was efficacious in inducing and maintaining remission compared with placebo in this study.15 16
Oral brepocitinib and ritlecitinib have completed phase II clinical studies for moderate to severe UC (VIBRATO umbrella study) with primary results suggesting significantly higher proportion of clinical remission and endoscopic improvement (p<0.05) with ritlecitinib 70 mg and 200 mg and brepocitinib 30 mg and 60 mg vs placebo at week 817 (table 1).
JAK-STAT inhibitors
Selective lymphocyte trafficking inhibitors
A new SC formulation of vedolizumab (VDZ) has been studied with the benefit of its convenient route of administration for patients who would like to avoid maintenance intravenous infusion therapy. SC administered VDZ was proven to be more effective than intravenous VDZ in Crohn’s patients for both induction and maintenance of remission. In two phase 3 RCTs, VISIBLE 1 and VISIBLE 2, 48.0% of patients receiving VDZ SC vs 34.3% of placebo group were in clinical remission at week 52 (p=0.008). Enhanced clinical response at week 52 was achieved by 52.0% vs 44.8% of patients receiving VDZ SC vs placebo, respectively (p=0.167). Steroid-free clinical remission was achieved in 45.3% of patients receiving VDZ SC vs 18.2% of placebo and 48.6% of anti-TNF-naive patients receiving VDZ SC vs 42.9% of those receiving placebo remained in clinical remission at week 52.
Injection site reaction was the only new safety finding observed for VDZ SC (2.9%). Similarly, in moderately to severely active UC patients, SC VDZ was effective as maintenance therapy in patients, who had a clinical response to intravenous VDZ induction therapy. It had demonstrated a favourable safety and tolerability profile as well. SC VDZ had also been shown to be drastically cost-effective. In TRAVELESS study, 124 patients agreed to transition from intravenous to SC VDZ. There were no statistically significant differences in disease activity scores between baseline and week 12. The most common adverse drug reaction reported was injection site reactions (15%). Based on this cohort, an expected reduction of £572 000 per annum was likely to be achieved.18–21
Etrolizumab is a gut selective monoclonal antibody (mAb) that selectively targets the β7 subunit of both α4β7 and α4E7 integrins.9 The HICKORY phase III study showed 105 mg 4 weekly etrolizumab-induced remission was significantly higher in patients with UC (18.5%) previously treated with anti-TNF agent at week 14 vs placebo (6.3%). There was no significant difference between groups in remission at week 62.22 In a double-blind RCT, the LAUREL study, no significant difference was noted between patients receiving 105 mg etrolizumab 4 weekly vs placebo at eeek 62 between those who reponed at Week 10.23 Similarly in GARDENIA study, in moderately to severely active UC patients, infliximab was used as an active comparator. Although the study did not show statistical superiority for the primary endpoint, etrolizumab performed similarly to infliximab from a clinical viewpoint and no significant difference was noted between patients receiving etrolizumab versus those who were receiving placebo.24
Carotegrast methyl (AJM300) is an oral small molecule inhibitor that targets the α4 subunit of α4β7 and α4β1. In a Japanese phase III double-blind RCT, AJM300 was well tolerated and induced a clinical response(defined as a reduction in Mayo Clinic score of 30% or more and 3 or more, a reduction in rectal bleeding score of 1 or more or rectal bleeding subscore of 1 or less, and an endoscopic subscore of 1 or less) in patients with moderately active UC who had an inadequate response or intolerance to mesalazine, at week 8. Patients receiving AJM300 960 mg three times per day had clinical response versus placebo, 46% vs 21%, respectively (p<0.0003) and endoscopic remission in 14% vs 3% of placebo (p<0.0057) The most common side effect was nasopharyngitis and no serious adverse effects were reported25 (table 2).
Novel selective lymphocyte trafficking inhibitors
IL12/IL23 inhibitors
Mirikizumab, an antibody against the p19 subunit of IL-23, was effective in UC; in a phase 2 RCT, at week 12, 15.9% (p=0.066), 22.6% (p=0.004) and 11.5% (p=0.142) of patients in the 50 mg, 200 mg and 600 mg groups, respectively, achieved clinical remission, cf 4.8% given placebo. Extended doses of mirikizumab (additional 12 weeks) produced a clinical response in up to 50% of patients who did not have a response post-12 weeks induction, most of whom maintained clinical response for up to 52 weeks.26 27 Another CD RCT showed statistically significant endoscopic and clinical response at week 12 for all mirikizumab groups versus placebo (200 mg: 25.8%, p=0.079; 600 mg: 37.5%, p=0.003; 1000 mg: 43.8%, p<0.001; Placebo: 10.9 %).26–28 Several phase 3 UC trials are ongoing including LUCENT 1 (NCT03518086) and LUCENT 2 (NCT03524092).
Risankizumab (RZB) (mAb against p19 subunit of IL23) in a phase II randomised, double-blind study, was more effective than placebo for inducing clinical remission in patients with CD who had failed ≥1 anti-TNF agent (31% vs 15% placebo, p<0.05) at week 12. A phase 2/3 trial for induction (NCT03398148) and a phase 3 study for maintenance treatment (NCT03398135) are ongoing in UC.29 30 In ADVANCE, MOTIVATE and FORTIFY randomised controlled phase III trials, at week 12, significantly greater proportions of patients with moderate to severe Crohn’s receiving RZB 600 mg intravenous achieved clinical remission (54.3% vs 23.8%) and endoscopic response (43.9% vs 13.3%) (p<0.001). At week 12, statistically higher proportions of RZB-treated patients achieved the composite endpoint CDAI clinical remission and endoscopic response, as well as endoscopic remission in the colonic only and ileal–colonic subgroups (p<0.001). At week 52, significantly greater proportions of patients receiving RZB 360 mg SC achieved clinical remission (54% vs 37.1%) and endoscopic response, in the colonic and ileal–colonic subgroups only (p≤0.05). In patients with endoscopic remission after 12 weeks of intravenous RZB sustained endoscopic remission at week 52 vs withdrawal in the colonic and ileal–colonic subgroups (p≤0.01). Results for ileal only CD at week 12 were not promising but were limited by the small number of patients in this subgroup.30 31
Most adverse effects were gastrointestinal in nature with a low rate of opportunistic infections. No deaths, malignancies, adjudicated major adverse cardiovascular events, latent/active tuberculosis or herpes zoster were reported. Treatment-emergent but none neutralising anti-drug antibodies were developed. One of the patients with maternal use of RZB preconception and during the first trimester had an elective miscarriage because of fetal defects (fetal cystic hygroma and hydrops fetalis) 78 days after RZB was discontinued, however, this was considered by the investigator as having a reasonable possibility of being related to RZB.30
With brazikumab (mAb against the p19 subunit of IL23) in a phase 2a double-blind, placebo-controlled CD trial, 49.2% achieved clinical remission cf 26.7% controls at week 8. The study group were patients with moderate to severe CD who failed anti-TNF treatment. Two infusions at weeks 0 and 4 achieved a higher rate of clinical response at week 8 compared with placebo and continued SC q4 weeks led to ongoing clinical response and remission at week 24. Brazikumab was well tolerated in this study but there was no endoscopic/imaging evaluation. The most common adverse events were headache and nasopharyngitis. Brazikumab is in a CD phase IIb/III (NCT03759288) and a UC phase II/open label extension trial (NCT03616821).32 33
Guselkumab is another human monoclonal antibody against the p19 subunit of IL23. A phase 2b/3, randomised, double-blind, placebo-controlled, parallel-group study (NCT04033445, QUASAR) is ongoing in patients with moderately to severely active UC anticipated completion by July 2025.
Combination therapy with guselkumab and golimumab is under investigation in a phase 2a randomised study (NCT03662542, VEGA-moderate to severe UC). GALAXI 1 is a phase II RCT for CD regarding guselkumab. Patients were randomised to guselkumab 200 mg, 600 mg or 1200 mg at weeks 0, 4 and 8; ustekinumab (reference arm) 6 mg/kg intravenous at week 0 and 90 mg SC at week 8; or placebo intravenous. There were significant decreases of Crohn's Disease Activity Index (CDAI) from baseline in all guselkumab groups cf placebo at week 12 and a higher proportion patients in all guselkumab groups who achieved clinical remission (200 mg: 54%, 600 mg: 56%, 1200 mg: 50%, placebo: 15.7%), clinical biomarker response (200 mg: 54%, 600 mg: 48%, 1200 mg: 42%, placebo: 11.8%) and endoscopic response (200 mg: 36%, 600 mg: 40%, 1200 mg: 36%, placebo: 11.8%). In biological failures, 45.5% (35/77) in the guselkumab group and 12.5% (3/24) with placebo achieved clinical remission at week 12. Rates of adverse effects, mainly infections were similar across all guselkumab groups cf placebo32 34 (table 3).
Novel IL-12/IL-23 inhibitors
S1P receptor modulators
S1P is a ligand of G protein coupled receptors (S1P1-S1P5) responsible for controlling the egress of lymphocytes from lymphoid organs. The S1P/S1PR interaction can also result in internalisation of the S1PR which leads to decrease lymphocyte release from the lymphoid tissue into the circulation.35
Ozanimod is a S1P receptor modulator with selective affinity for S1PR1 and S1PR5. A phase III RCT in UC used oral 1 mg Ozanimod in a 10-week induction period with responders followed up to week 52. Incidence of clinical remission was significantly higher among ozanimod group than placebo during induction (18.4% vs 6.0%, p<0.001) and maintenance (37.0% vs 18.5%, p<0.001). Clinical response was significantly higher with ozanimod than placebo during induction (47.8% vs 25.9%, p<0.001) and maintenance (60.0% vs 41.0%, p<0.001). Incidence of infection with ozanimod was similar to placebo during induction and higher than placebo during maintenance. Serious infection occurred in <2%. Elevated liver aminotransferase levels were more common with ozanimod.36–38 Ozanimod has been approved for treatment of moderate to severe UC by FDA and European Commission. In stepstone phase II prospective single-arm CD trial, clinical remission was shown in 39.1%, and response in 56.5% of patients at week 12. Endoscopic and histological improvements were also seen within 12 weeks of initiating ozanimod therapy. Phase 3 placebo-controlled trials have been initiated.39
Etrasimod is an oral S1P receptor modulator with selectivity for S1PR1, S1PR4 and S1PR5. In a phase II UC trial, etrasimod 2 mg showed significantly higher clinical remission vs placebo (33% vs 8.1% of placebo, p<0.001) and endoscopic improvement (41.8% vs 17.8% of placebo, p<0.003). Phase III UC trials are currently recruiting (NCT03996369, NCT03945188, NCT03950232, NCT04176588). A phase II/III CD study is currently recruiting (NCT04173273). Most common adverse reactions were disease worsening, upper respiratory tract infections, nasopharyngitis and anaemia (table 4).32 40
Novel S1P-Receptor modulators
Phosphodiesterase 4 inhibitors
Phosphodiesterases (PDE1-PDE11) are a group of intracellular enzymes that catalyse the breakdown of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate. PDE4 catalyses the breakdown of cAMP in multiple cells including T-cells, macrophages and monocytes leading to activation of nuclear transcription factor kappa B (NF-κB), promoting proinflammatory effects. Inhibition of PDE4 can lead to suppression of NF-κB, reducing TNF-α mRNA expression and production of nitric oxide and increasing synthesis of anti-inflammatory cytokines.32
Apremilast is an oral small-molecule PDE 4 (PDE4) inhibitor. A phase II RCT involving patients with active UC had shown clinical remission at week 12 by 31.6% of patients in the 30 mg apremilast group and 12.1% of patients in the placebo group (p<0.01). However, only 21.8% with 40 mg apremilast achieved clinical remission at week 12 (p<0.27). Although the primary endpoint of clinical remission was not met, a greater proportion of patients receiving apremilast had improvements in clinical, endoscopic and inflammatory markers at 12 weeks. Most frequent apremilast-associated adverse events were headache and nausea.41
Toll-like receptor 9 inhibitor
Cobitolimod is an oligodeoxynucleotide that binds to Toll-like receptor 9 (TLR9) on lymphocytes and antigen presenting cells. It activates TLR 9 leading to induction of regulatory T-cells that produce anti-inflammatory IL-10 in addition to suppressing proinflammatory TH-17 cells. Cobitolimod is a topical agent with low systemic absorption.32 A phase IIb RCT (CONDUCT) randomised patients with moderate to severe left-sided UC. The primary endpoint of clinical remission at week 6 was met by the 250 mg of cobatolimod group with 21% vs 7% in the placebo group (p<0.025).42
Micro-RNA-124 agonist
ABX464 is an oral agent that binds with the cap binding complex, allowing specific splicing of anti- inflammatory miR-124 (microRNA-124), modulating the proinflammatory cytokines. A phase II RCT in patients with UC treated with 50 mg of ABX464 found at week 8, 35.0% and 70.0% of patients in the achieved clinical remission and clinical response vs 11.1% and 33.3% in the placebo group. Endoscopic improvement and remission were observed in 50.0% and 10.0% of patients receiving ABX464, respectively, vs 11.1% each for placebo. Maintenance therapy with ABX464 sustained remission and brought additional patients into remission. The trial showed a good safety profile with headache to be the most common adverse effect reported. Phase II trials for CD were announced43 (table 5).
Novel categories in the pipeline
Anti-TNF agents
A SC formulation of infliximab has recently been shown to be as effective as intravenous infliximab. A recent multicentre cohort study with 181 patients with IBD, majority with Crohn’s already on 8-weekly dosing of intravenous infliximab prior to switching and more than half (59.1%) on concomitant immunomodulatory therapy had shown no significant difference between baseline and repeat measurements at 3, 6 or 12 months for HBI, SCCAI, C reactive protein or FC. IN SC group, median infliximab level increased from a baseline of 8.9 µg/dL (range 0.4–16) to 16.0 µg/dL (range 2.3–16, p<0.001) at 3 months and serum levels stayed stable at 6 months (median 16 µg/dL, range 0.3–17.2) and 12 months (median 16 µg/dL, range 0.3–19.1, both p<0.001 compared with baseline). Treatment persistence, patient acceptance and satisfaction rates with SC CT-P13 were also very high. What is more, patients with perianal CD who were switched from intravenous to SC IFX had high rate of symptom free survival and treatment persistence at 6 months, with comparable efficacy and safety with intravenous Infliximab at 6 months.44 45
Conclusion
In the new COVID-19 era, immunosuppression has become a serious concern for most of our patients and optimising their treatment with tailored therapies has become a necessity to optimise the balance between treating their disease effectively and their concerns, regarding their immunosuppression and vulnerability. Medical practitioners should develop individualised treatment plans based on a comprehensive assessment of the patient. The treatment should be flexible and changed according to the patient’s response. Timely communication and close cooperation between doctors and patients are equally essential to effective treatment strategies.
The new ‘treat to target approach’ although cost-effective and difficult to achieve, has also identified a considerable number of patients who lost response after their treatment. Optimisation of IBD treatment also involves moving to a new category of oral agents in the future, that will not require hospitalisation that is not only stressful for many of our patients but also cost-demanding.9
Although, more clinical data are required on long-term safety, with deepening of the research around IBD pathogenesis, new therapies are coming into view, with a safer adverse effect profile and promising results not only on patients with IBD with mild disease but also for those who are failing one or more biological agents and those who would like to avoid a colectomy at any cost. We still confront with many unresolved challenges regarding efficacy versus safety and more long-term data are required to offer targeted therapies that minimise the risk and optimise outcomes.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors Both authors contributed equally to writing and reviewing the document.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.
Linked Articles
- UpFront